Home > Organization > Divisions and Independent Research Units > Division of Cancer Therapeutics > Research Highlights
Research Highlights
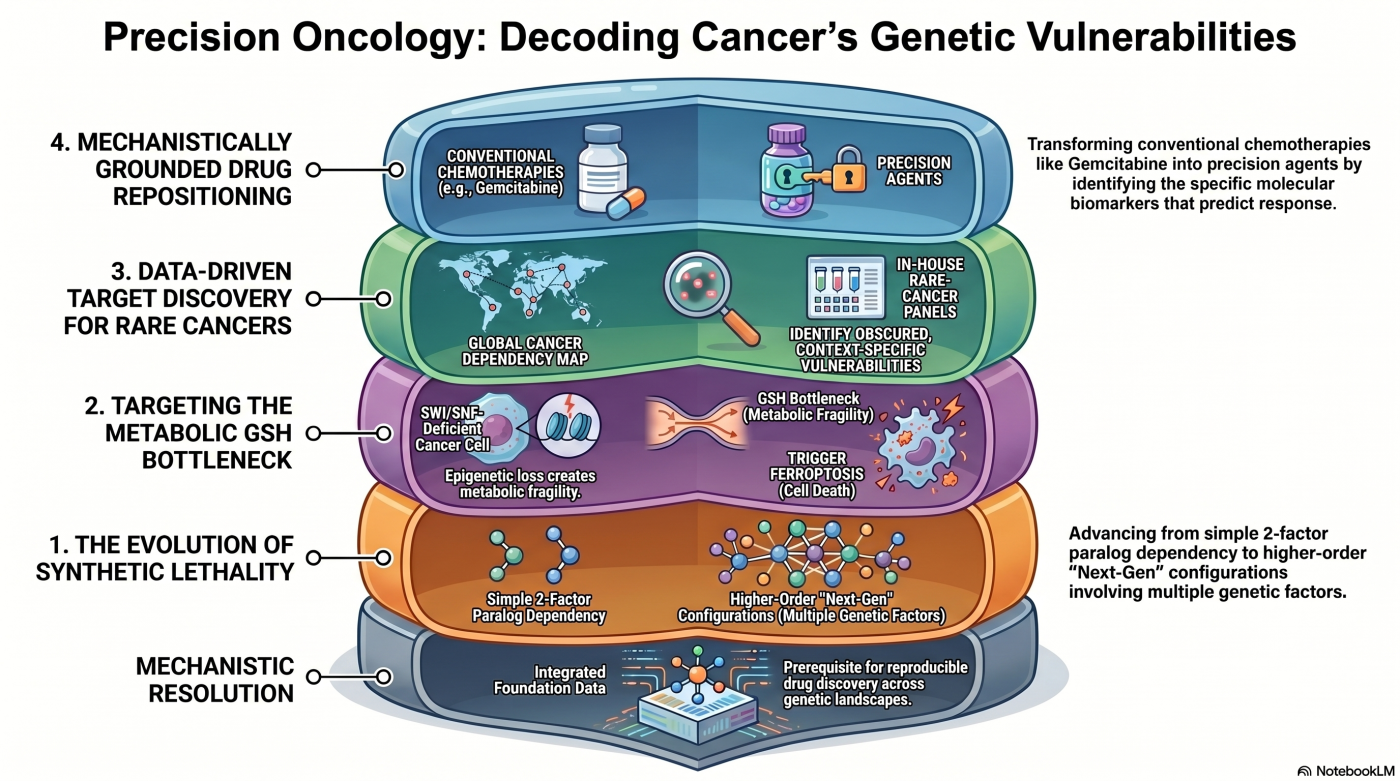
Mechanistic Resolution Across Six Research Themes
This page presents the laboratory's six research themes as a coherent narrative, not a catalog. Each theme reflects the same commitment: resolving which molecule and pathway define a cancer cell's vulnerability, and how that vulnerability can be therapeutically exploited. That mechanistic resolution is our prerequisite for reproducible drug discovery.
The six themes trace two arcs. Themes 1 through 3 chart the conceptual evolution of the laboratory's synthetic-lethality strategy: from the two-factor paralog-dependency framework (Theme 1 — Conventional Synthetic Lethality), through the three-factor paralog co-inhibition framework (Theme 2), to higher-order multi-factor configurations (Theme 3 — Next-Generation Synthetic Lethality). Themes 4 through 6 represent three parallel axes of target discovery: data-driven identification from public dependency databases (Theme 4), metabolic vulnerability in SWI/SNF-deficient cancers (Theme 5), and mechanistically grounded repositioning of approved drugs (Theme 6).
Quick Navigation:
- Theme 1 — Conventional Synthetic Lethality
- Theme 2 — Paralog Co-Inhibition
- Theme 3 — Next-Generation Synthetic Lethality
- Theme 4 — Data-Driven Target Discovery
- Theme 5 — Glutathione Metabolic Vulnerability
- Theme 6 — Drug Repositioning
Theme 1: Conventional Synthetic Lethality
Stage: Mechanistic Validation | Preclinical Validation
The Two-Factor Paralog-Dependency Foundation
Ogiwara et al., Cancer Discov. 2016 — Oike et al., Cancer Res. 2013
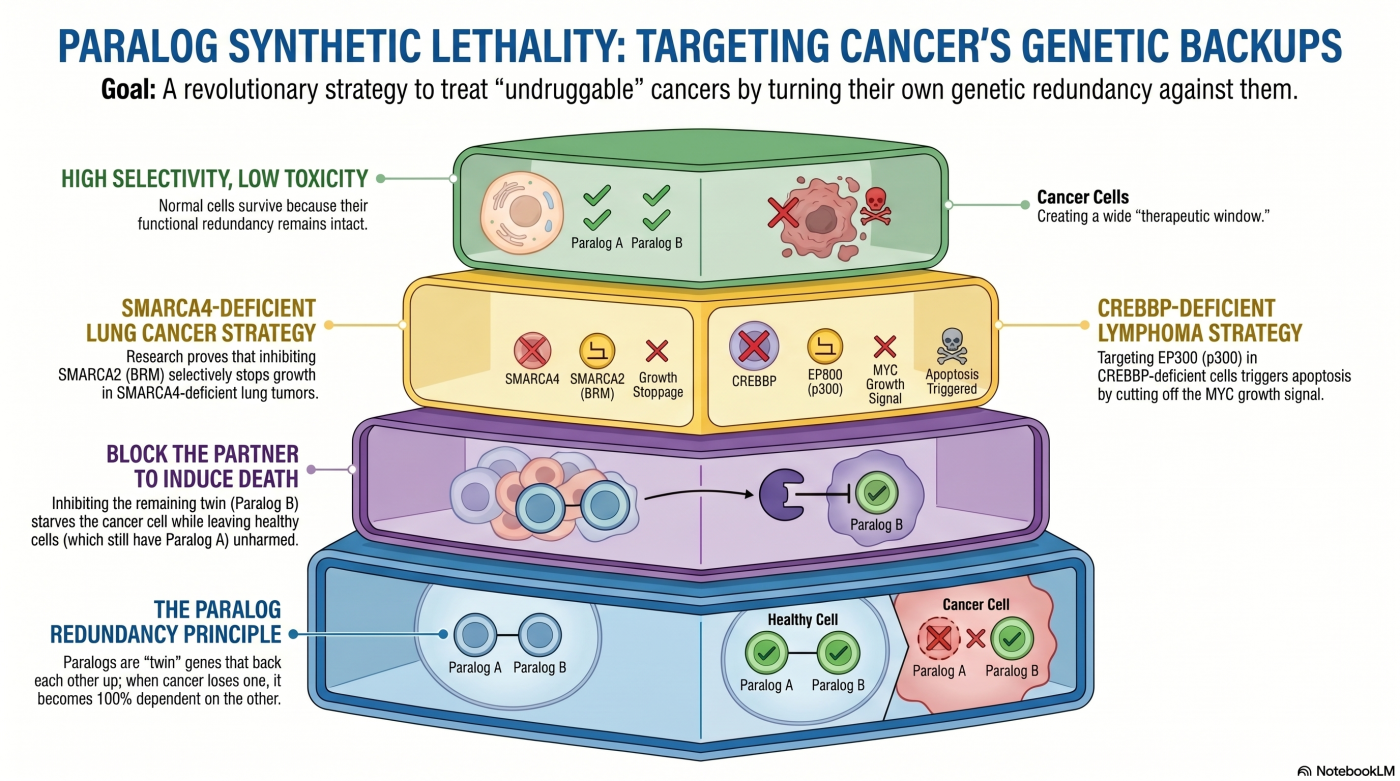
Synthetic lethality describes a vulnerability that emerges only when two genetic perturbations coexist in the same cell. The simplest and most studied form — the two-factor case — is paralog dependency: when one member of a functionally redundant paralog pair is lost in cancer, the surviving paralog can become a critical dependency in that cancer cell specifically. Inhibiting the survivor selectively impairs the cancer cell because the functional redundancy that would normally buffer the perturbation has already been consumed by the driver mutation.
We demonstrated this principle across two independent paralog pairs in two cancer contexts. In SMARCA4 (BRG1)-deficient non-small cell lung cancer, the paralogous ATPase SMARCA2 (BRM) functionally compensates for SWI/SNF chromatin remodeling in SMARCA4-deficient cells. siRNA-mediated BRM suppression induced cellular senescence and arrested tumor growth in BRG1-deficient cells, with selectivity over BRG1-intact cells confirmed both in cell culture and mouse xenograft models (Oike et al., Cancer Res 2013).
Three years later, we reported the CBP–p300 axis in CBP (CREBBP)-deficient cancers — small-cell lung cancers and B-cell lymphomas (DLBCL and follicular lymphoma) — showing that CBP loss creates increased dependency on the paralog p300 (EP300) to maintain enhancer-driven transcription of MYC (Ogiwara et al., Cancer Discov 2016). The research-use p300 inhibitor C646 abrogated H3K27ac at the MYC super-enhancer, collapsed MYC expression, and preferentially induced apoptosis in CBP-deficient cells. The cell-line panels and conceptual architecture established here became the substrate for the metabolic-vulnerability studies (Theme 5) and data-driven discovery work (Theme 4), and ultimately the three-factor paralog co-inhibition strategy (Theme 2).
(Figure placeholder — replace with figure or remove before publication)
Key Publications:
- Ogiwara H et al. Cancer Discov. 2016;6(4):430–445. → PubMed
- Oike T et al. Cancer Res. 2013;73(17):5508–5518. → PubMed
→ Related: Paralog Co-Inhibition — the conceptual successor; Research Projects.
Theme 2: Paralog Co-Inhibition
Stage: Preclinical Validation | Drug-Discovery Opportunity
A Three-Factor Synthetic Lethality Strategy
Sasaki et al., Nat Commun. 2024 — Sasaki et al., Cancer Res Commun. 2025
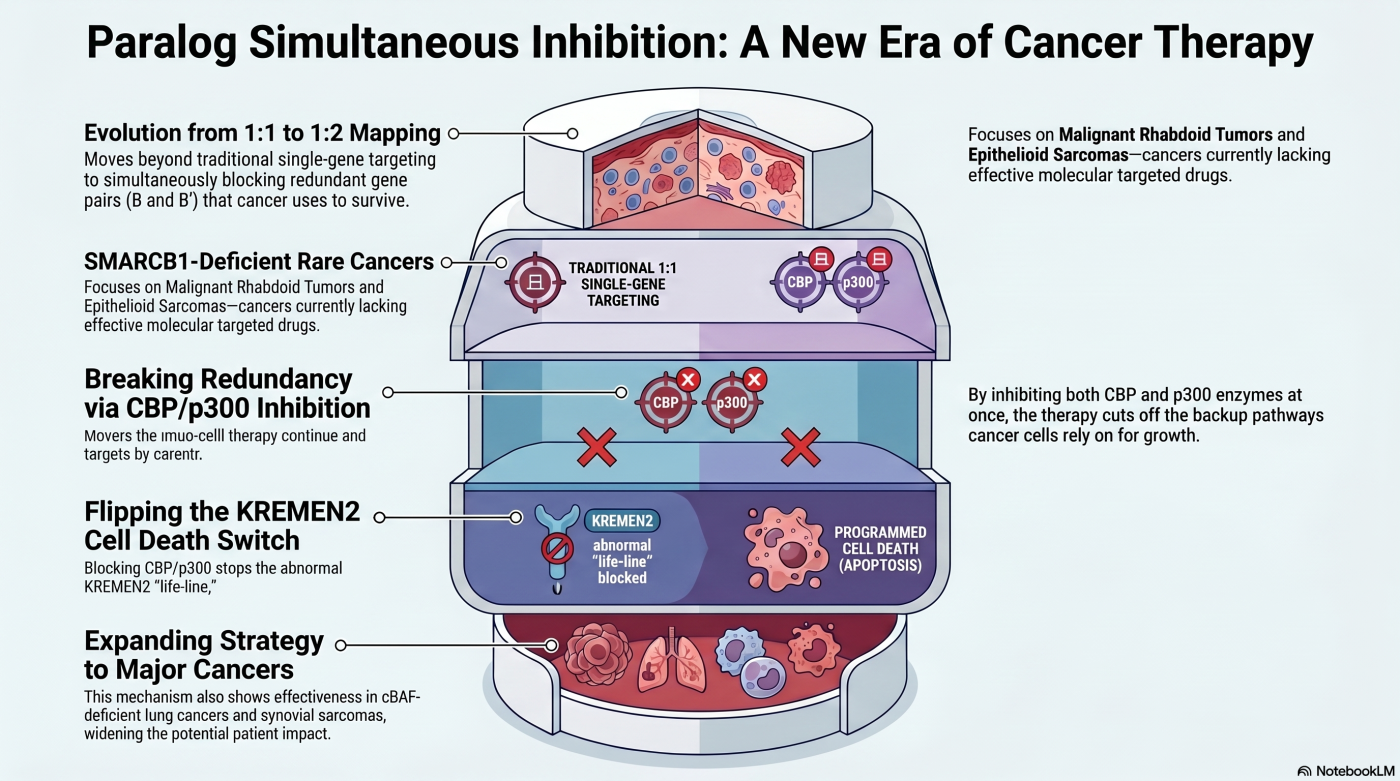
Conventional paralog-dependency synthetic lethality operates between two factors: one gene lost in cancer, and one surviving paralog as the therapeutic target. Approximately one-third of human genes have functional paralogs whose redundancy buffers single-gene loss. In cases where the cancer-driver gene and its therapeutic target are themselves a redundant paralog pair, inhibiting only one paralog may allow the other to compensate. We extended synthetic lethality to a three-factor framework — pioneered in our laboratory — by designing a strategy that simultaneously inhibits both members of a paralog pair that would otherwise buffer each other.
We first established this framework against SMARCB1-deficient rare cancers, including malignant rhabdoid tumor (a pediatric solid tumor with near-universal SMARCB1 loss) and epithelioid sarcoma (a rare AYA soft-tissue sarcoma), by simultaneously inhibiting the histone acetyltransferase paralog pair CBP and p300 (Sasaki et al., Nat Commun 2024). SMARCB1 loss removes cBAF-mediated repression of the gene KREMEN2, and KREMEN2 becomes a critical survival-associated transcriptional output in SMARCB1-deficient cells. Dual CBP/p300 inhibition collapses KREMEN2 transcription, releases pro-apoptotic KREMEN1, and preferentially induces cell death in SMARCB1-deficient cells. The publication-disclosed research compound CP-C27 (dual CBP/p300 inhibitor) validated the mechanism in vitro and in vivo. The framework was subsequently extended to cBAF-deficient cancers more broadly — SMARCA4-deficient non-small cell lung cancer and SS18-SSX-driven synovial sarcoma — demonstrating that the strategy tracks the molecular alteration across histologies rather than being tissue-restricted (Sasaki et al., Cancer Res Commun 2025).
(Figure placeholder — replace with figure or remove before publication)
Key Publications:
- Sasaki M et al., Ogiwara H. Nat Commun. 2024;15(1):4770. → PubMed
- Sasaki M et al., Ogiwara H. Cancer Res Commun. 2025;5(1):24–38. → PubMed
→ Related: Conventional Synthetic Lethality — the conceptual origin; Next-Generation Synthetic Lethality — the higher-order expansion; Research Projects.
Theme 3: Next-Generation Synthetic Lethality
 Stage: Target Discovery | Preclinical Validation
Stage: Target Discovery | Preclinical Validation
Higher-Order Synthetic Lethality — 2-to-1 and Beyond
Takeuchi et al., NPJ Precis Oncol. 2026 (in press)
Next-generation synthetic lethality — or, in technical contexts, higher-order synthetic lethality — describes synthetic-lethal interactions that involve more than two genetic factors: for example, a 2-to-1 configuration in which two driver gene losses together create dependence on a single therapeutic target. The two-factor and three-factor frameworks of Themes 1 and 2 share an implicit assumption that at least one member of the synthetic-lethal pair remains present and targetable. Higher-order configurations address cancer genotypes where this assumption fails.
Dual SMARCA4/SMARCA2-deficient cancers — a subset of lung adenocarcinomas, small cell carcinoma of the ovary hypercalcemic type (SCCOHT), and thoracic SMARCA4-deficient undifferentiated tumors — have lost both core SWI/SNF ATPase subunits. The conventional SMARCA2-targeting strategy (Theme 1) is inapplicable because SMARCA2 itself is absent. We asked whether cancers that have eliminated the entire SWI/SNF ATPase function develop a critical compensatory dependency on alternative chromatin-remodeling complexes.
A siRNA screen targeting 17 ATP-dependent chromatin remodeling factors in dual SMARCA4/SMARCA2-deficient versus isogenic SMARCA4-reconstituted cell lines identified CHD3 — the core ATPase of the CHD3/NuRD nucleosome-remodeling complex — as a selective synthetic-lethal dependency. Validation across a broad cell-line panel confirmed that dual loss is the determinant: SMARCA4 single-deficient lines show an intermediate response; CHD4, the close CHD3 paralog, does not reproduce the synthetic lethality.
Mechanistically, CHD3/NuRD serves as an "epigenetic brake" at the PARD3B enhancer under conditions of SWI/SNF loss. Combined RNA-seq, ATAC-seq, and CUT&RUN analyses showed that CHD3 constitutively maintains chromatin closure at the PARD3B locus; CHD3 inhibition releases this brake, drives PARD3B de-repression, and attenuates MYC-pathway transcriptional output, selectively impairing dual-deficient cells. Rescue by co-knockdown of CHD3 and PARD3B confirmed PARD3B as the causal mediator. This is a gain-of-toxicity mode of synthetic lethality — cell death arises from the toxic de-repression of a normally silenced factor, not from the loss of a survival signal. In vivo, doxycycline-inducible CHD3 ablation in dual-deficient SBC-5 CDX models produced potent tumor regression. Analysis of TCGA lung adenocarcinoma cases showed that SMARCA4-mutant tumors display significantly lower PARD3B expression, supporting the epigenetic brake model in human tumor data.
This work introduces two new conceptual frameworks: cross-complex dependency (functional dependency between distinct chromatin-remodeling complexes) and the gain-of-toxicity mode of higher-order synthetic lethality. Because the epigenetic brake depends on the structural integrity of CHD3/NuRD rather than its HDAC enzymatic activity, protein-degradation-based approaches may provide one potential route for targeting CHD3/NuRD dependency.
(Figure placeholder — replace with figure or remove before publication)
Key Publication:
- Takeuchi M, Okimoto Y, Fukushima M, Hirano H, Ogiwara H*. NPJ Precis Oncol. 2026 (in press). [PubMed link to be added when available]
→ Related: Conventional Synthetic Lethality; Paralog Co-Inhibition; Research Projects.
Theme 4: Data-Driven Target Discovery
Stage: Preclinical Validation
Reading Public Dependency Data Through a Laboratory-Specific Lens
Saito et al., NPJ Precis Oncol. 2025
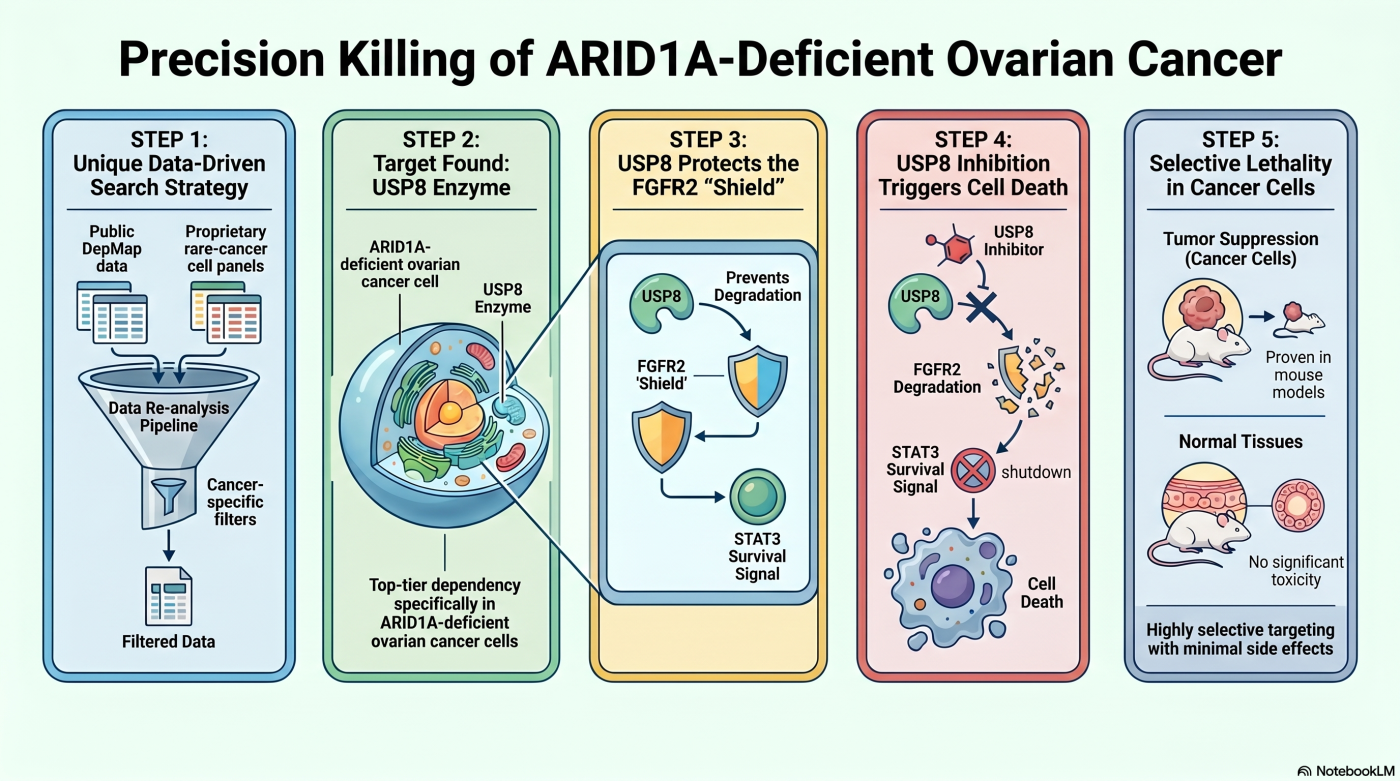
Public cancer dependency datasets offer a broad signal, but not the right resolution for rare and disease-specific vulnerabilities. Our data-driven target discovery pipeline integrates the Cancer Dependency Map (DepMap) with in-house rare-cancer cell-line panels, applying three analytical layers: (1) context-specific re-analysis restricted to a single cancer lineage rather than pan-cancer averaging, (2) integration with in-house cell lines absent from DepMap, including Japan-specific and rare cancer subtypes, and (3) pathway-level interpretation accounting for paralog-mediated functional redundancy that can suppress dependency signals.
The representative output of this pipeline is the identification of USP8 — a deubiquitinating enzyme (DUB) — as a synthetic-lethal dependency in ARID1A-deficient ovarian clear cell carcinoma (OCCC) (Saito et al., NPJ Precis Oncol 2025). Ovarian clear cell carcinoma is a histological subtype markedly more prevalent in Japanese women than in Western populations; approximately 50% of cases carry ARID1A loss-of-function mutations, and treatment options at recurrence are limited.
Restricting DepMap re-analysis to ovarian-lineage cell lines stratified by ARID1A status surfaced USP8 as a top-ranked differential dependency — a signal obscured by pan-cancer averaging. We validated this prediction by CRISPR/Cas9 screening in our in-house OCCC cell-line panel, with results compared against Chronos-based DepMap dependency scores. Mechanistically, USP8 deubiquitinates and stabilizes FGFR2; ARID1A-deficient cells develop increased dependency on the resulting FGFR2–STAT3 survival axis. USP8 inhibition triggers FGFR2 degradation, collapses STAT3 signaling, and preferentially induces cell death in ARID1A-deficient cells — an effect reproduced in mouse xenograft models. The result anchors a precision-medicine framework where a single molecular biomarker (ARID1A loss) prospectively identifies the responsive population.
(Figure placeholder — replace with figure or remove before publication)
Key Publication:
- Saito R et al., Ogiwara H. NPJ Precis Oncol. 2025;9(1):69. → PubMed
→ Related: Conventional Synthetic Lethality; Research Projects.
Theme 5: Glutathione Metabolic Vulnerability
Stage: Preclinical Validation | Drug-Discovery Opportunity
Epigenetic Loss, Metabolic Bottleneck, Ferroptotic Cell Death
Takeuchi et al., Cancer Res. 2026 — Ogiwara et al., Cancer Cell. 2019 — Sasaki et al., Sci Rep. 2024
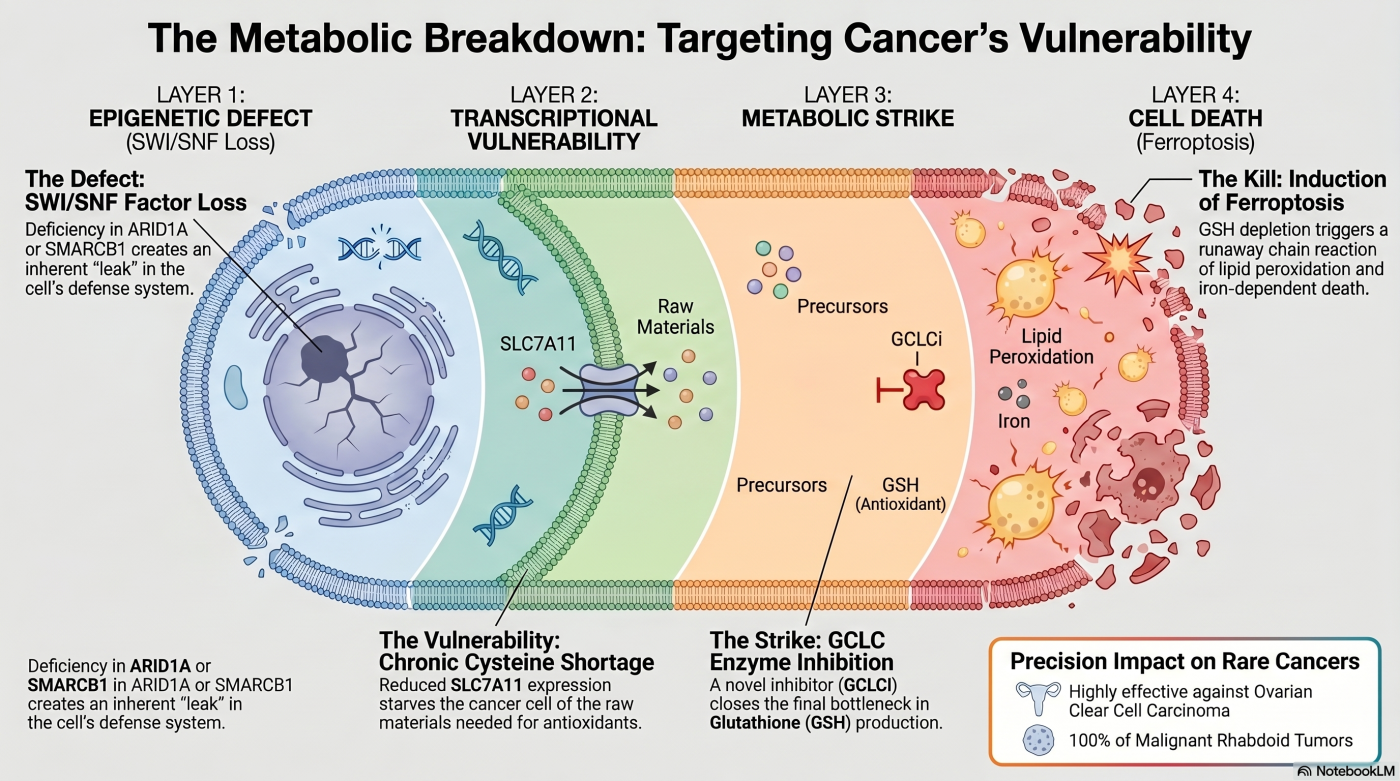
Synthetic lethality extends beyond gene-gene relationships to encompass the metabolic fragility created by epigenetic dysregulation. This theme traces a four-layer causal chain: epigenetic alteration → transcriptional silencing → metabolic bottleneck → iron-dependent cell death (ferroptosis).
We first reported that ARID1A-deficient cancers depend on glutathione (GSH) metabolism (Ogiwara et al., Cancer Cell 2019). ARID1A normally activates transcription of the cystine transporter SLC7A11; ARID1A loss silences SLC7A11, chronically restricts cysteine uptake, and creates a GSH biosynthesis bottleneck. These cells are consequently hypersensitive to GSH-depleting agents. Subsequent work generalized the SLC7A11-mediated vulnerability to the broader SWI/SNF-deficient landscape — including SMARCA4 and PBRM1 loss — across multiple cancer types (Sasaki et al., Sci Rep 2024).
In 2026, we extended this line to SMARCB1-deficient rare cancers — malignant rhabdoid tumor and epithelioid sarcoma — by targeting GCLC (glutamate–cysteine ligase catalytic subunit), the rate-limiting enzyme of GSH biosynthesis (Takeuchi et al., Cancer Res 2026). Novel GCLC inhibitors GCLCi1 / GCLCi0, developed in collaboration with Ono Pharmaceutical Co., Ltd. (disclosed in the publication), preferentially reduce intracellular GSH levels in SMARCB1-deficient cells. GSH depletion inactivates GPX4, allowing iron-catalyzed lipid peroxidation to proceed and inducing ferroptosis (iron-dependent cell death). The mechanism was validated by ferrostatin-1 rescue, direct GSH measurement, and lipid-peroxidation quantification. GCLCi compounds showed higher SMARCB1-loss-vs-intact selectivity than tazemetostat in the in vitro epithelioid sarcoma models tested, and suppressed tumor growth in xenograft models, with additive efficacy in combination with the glutaminase inhibitor telaglenastat.
(Figure placeholder — replace with figure or remove before publication)
Key Publications:
- Takeuchi M et al., Ogiwara H. Cancer Res. 2026. (GCLC inhibitors and ferroptosis in SMARCB1-deficient rare cancers.) → PubMed
- Ogiwara H et al. Cancer Cell. 2019;35(2):177–190.e8. (Glutathione metabolic dependency in ARID1A-deficient cancers.) → PubMed
- Sasaki M, Ogiwara H. Sci Rep. 2024;14(1):31321. (Generalization across the SWI/SNF-deficient landscape.) → PubMed
→ Related: Paralog Co-Inhibition; Research Projects.
Theme 6: Drug Repositioning
Stage: Preclinical Validation | Retrospective Clinical Evidence
Mechanistically Grounded Repositioning of Gemcitabine
Hirano et al., Mol Cancer Res. 2026 — Kuroda et al., Gynecol Oncol. 2019
Existing drugs can provide translational opportunities when their biomarker-defined sensitivity mechanisms are understood. This theme demonstrates how a defined molecular mechanism can transform a conventional chemotherapeutic into a candidate for biomarker-guided repositioning — in a molecularly characterized patient population.
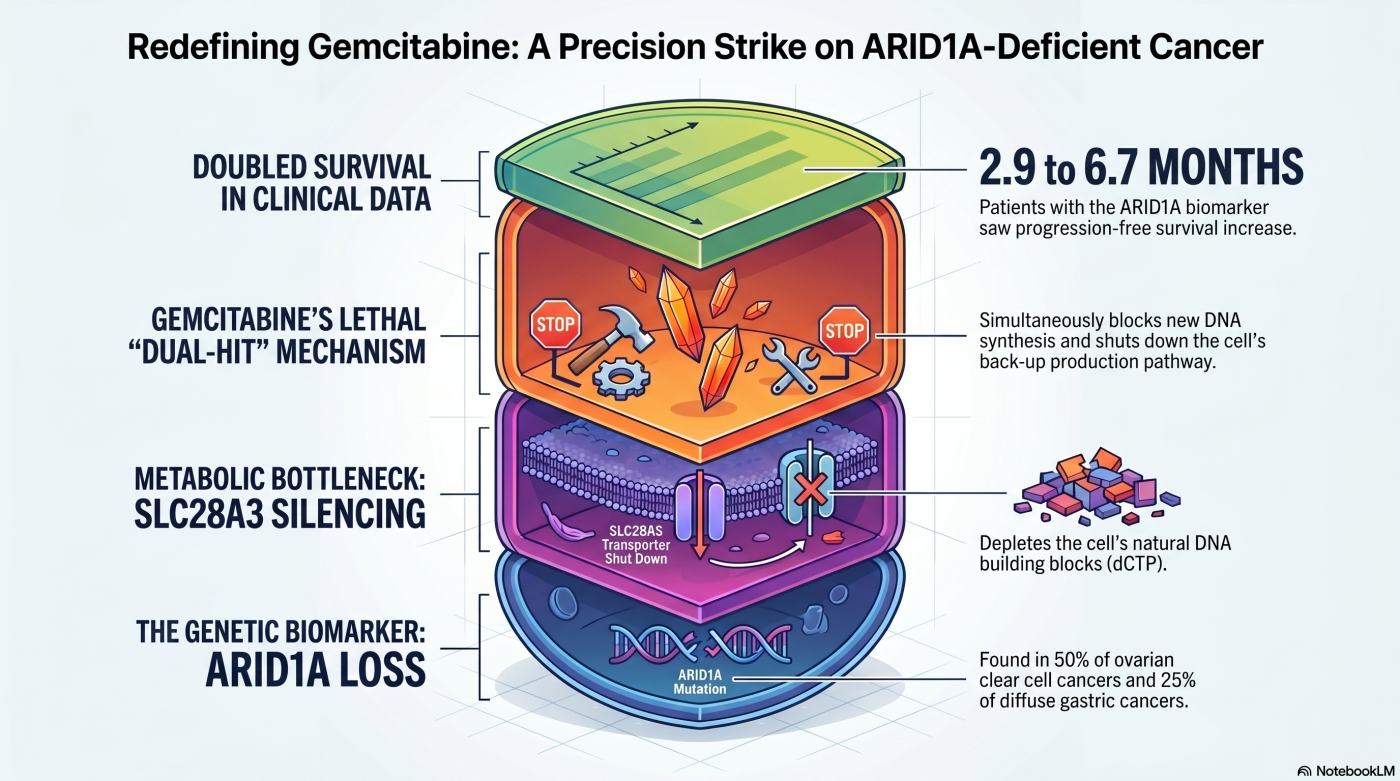
We first reported, by comparative drug sensitivity screening across ovarian clear cell carcinoma cell-line panels, that ARID1A-deficient cells are substantially more sensitive to gemcitabine than ARID1A-intact cells. A retrospective analysis of 149 ovarian clear cell carcinoma patients at the National Cancer Center and affiliated hospitals showed that progression-free survival on second-line gemcitabine monotherapy was longer in ARID1A-deficient cases (median 6.7 months vs. 2.9 months; P = 0.02) (Kuroda et al., Gynecol Oncol 2019). The molecular basis was unresolved at that stage.
In 2026, we resolved the mechanism in ARID1A-deficient diffuse-type gastric cancer, including scirrhous gastric cancer (Hirano et al., Mol Cancer Res 2026). ARID1A loss silences the high-affinity nucleoside transporter SLC28A3, chronically depleting the intracellular dCTP pool. Gemcitabine then exerts a dual-hit effect in these cells: its triphosphate (dFdCTP) incorporates into DNA and induces chain termination, while its diphosphate (dFdCDP) inhibits ribonucleotide reductase (RNR) and blocks de novo dCTP synthesis. Normally, cells rescue this by importing deoxycytidine via SLC28A3 — the escape route ARID1A-deficient cells have already lost. All three dCTP-supply tracks are simultaneously impaired, driving selective cell death. The mechanism was validated by metabolomics, transcriptomics, ARID1A-rescue experiments, and tumor-growth inhibition in a peritoneal-dissemination xenograft model.
This work provides a mechanistic basis for biomarker-guided repositioning of gemcitabine in ARID1A-deficient cancers across two disease contexts sharing a single genetic denominator.
(Figure placeholder — replace with figure or remove before publication)
Key Publications:
- Hirano H et al., Ogiwara H. Mol Cancer Res. 2026. (Pyrimidine-metabolism vulnerability and dual-hit selective cell death in ARID1A-deficient diffuse gastric cancer.) → PubMed
- Kuroda T et al., Ogiwara H. Gynecol Oncol. 2019;155(3):489–498. (ARID1A-deficient OCCC retrospective cohort, n = 149.) → PubMed
→ Related: Data-Driven Target Discovery — ARID1A-deficient OCCC in the same genetic context; Research Projects.
Research Stage
All findings described on this page represent basic or preclinical research, or retrospective clinical data analysis. Gemcitabine (Theme 6) is an approved drug, but the biomarker-based patient-selection strategy described here has not been established as standard clinical practice. GCLCi1/GCLCi0 (Theme 5) and CP-C27 (Theme 2) are research-stage compounds disclosed in the corresponding publications. All other therapeutic strategies described on this page are in the preclinical research stage. Clinical translation of any target or strategy requires further preclinical validation, safety and pharmacokinetic evaluation, and clinical trials. These findings do not constitute clinical treatment recommendations.
Related Pages
- Research Projects — full platform architecture, disease-focused programs, and partnership inquiries
- Publications — complete publication record and Selected Publications Overview by theme
- Lab Members — laboratory team and research environment
- Home — laboratory overview and six-theme summary
- Contact and Access — collaboration, academic, and media inquiries
Last Updated: 2026-05-19
