約70%症例でみられる高頻度の遺伝子異常を発見 日本人に特徴的な発がん要因の存在も初めて確認日本人症例を中心とした大規模な肝細胞がんゲノムを解読
独立行政法人 国立がん研究センター
国立大学法人 東京大学
国立がん研究センター(理事長:堀田知光)と東京大学(総長:濱田純一)は、米国ベイラー医科大学と共同で、日本人を中心とする肝細胞がん症例608例のゲノム解読を行い、新たな治療標的候補の同定、また日本人に特徴的な発がん要因の存在の推定に成功しました。
本研究は、これまでで最大の肝細胞がんゲノム解析であり、重要なゲノム異常(ドライバー遺伝子:用語説明1)を最も高精度に同定したもので、今後、日本人における肝細胞がんの治療開発推進に重要な貢献をするものと考えられます。
本研究では最新の解析技術を駆使することで、同じ肝細胞がんであってもそれぞれの人種背景によってその原因となる因子の組み合わせが異なること、さらに日本人において特徴的で、これまで重要な発がん危険因子として知られている肝炎ウイルス感染とは異なる、未知の肝細胞がんの発がん要因が存在する可能性を世界でも初めて明らかにしました。また、がんゲノム解読データを元に演繹的に既知の発がん因子との関連や新たな原因を探索する研究は極めて新しい研究分野であり、近年、日本においても漸増している非肝炎ウイルス性肝細胞がんの治療・予防への貢献も強く期待されます。
この研究は、国立がん研究センター研究所 がんゲノミクス研究分野長・東京大学医科学研究所附属ヒトゲノム解析センター ゲノムシークエンス解析分野 教授 柴田龍弘、東京大学先端科学技術研究センターゲノムサイエンス教授 油谷浩幸らによる第3次対がん総合戦略研究推進事業「国際協調に基づく日本人難治がんゲノムデータベースの構築(国際がんゲノムコンソーシアム:International Cancer Genome Consortium、以下ICGCプロジェクト)」により行われたものです。また、本研究成果は国際科学誌「Nature Genetics」に2014年11月2日付(日本時間11月3日)オンライン版で発表されました。
研究成果
解析1
肝細胞がんの日本人413例並びに米国人90例(合計503例)のがん組織・正常組織について、高速シークエンサー(用語説明2)を用いた全エクソン解読並びに一部の症例では全ゲノム解読(用語説明3)を行い、得られた情報を東京大学医科学研究所附属ヒトゲノム解析センターの大型計算機(スーパコンピューター)を用いて解析しました。
遺伝子異常である30個のドライバー遺伝子を同定、中でもテロメラーゼ遺伝子異常が最も高頻度(約70%)であることを発見
この解析の結果、肝細胞がんの発生に重要なドライバー遺伝子を30個同定し(今回新たに同定した13個を含む)、さらにそれらの遺伝子が組み込まれた11の分子経路を見出しました(図1)。また中でも、テロメラーゼ遺伝子(用語説明4)が肝細胞がんにおいて最も高頻度なドライバー遺伝子であることを確認しました。細胞のがん化において重要な異常であるテロメラーゼ遺伝子の活性化に繋がる異常として、テロメラーゼ遺伝子の発現調節領域(プロモーター)の活性型突然変異、テロメラーゼ遺伝子のコピー数増加、B型肝炎ウイルス挿入が相互排他的に起こり、かつ肝細胞がん全体の約70%でこれらの異常のいずれかが起こっていることを発見しました。テロメラーゼ遺伝子異常の頻度は肝炎ウイルスの種類(B型肝炎、C型肝炎、非ウイルス性)とは無関係であり、日本人、米国人いずれのグループでも70%程度で異常を認め、これまで肝細胞がんで発見されたゲノム異常の中で最も頻度が高い重要なものであることが分かりました。
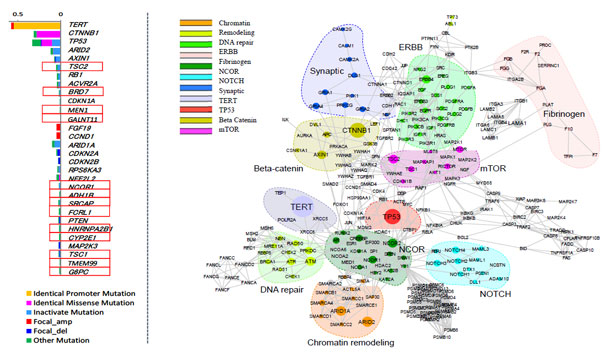
図1:
左 今回の解析で同定した肝細胞がんにおける30個のドライバー遺伝子。赤枠は今回新たに発見した13個のドライバー遺伝子
右 肝細胞がんにおいて異常を来している11の分子経路
分子標的候補の発見と既存阻害剤の有効性を示唆
同定した30個のドライバー遺伝子の中にはPIK3CA/mTOR経路(用語説明5)の活性化を来す遺伝子異常を複数認め、全体の45%の症例でPIK3CA/mTOR経路に何らかの異常が認められました(図2)。さらに、細胞株を使った検討からこうしたゲノム異常をもった肝細胞がん細胞に対して、分子標的薬であるmTOR阻害剤が有効である可能性が示唆されました。新たなドライバー遺伝子としてエピゲノム制御分子や細胞代謝関連酵素も複数同定し、また頻度は低いものの分子標的薬がすでに開発されているFGFR2, KIT, JAK1, METといった遺伝子の異常も確認しました。
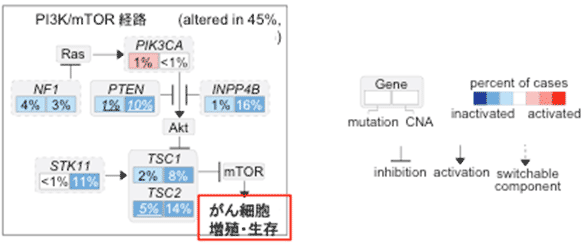
図2 肝細胞がんにおけるPIK3CA/mTOR経路の異常
解析2
解析1の503例に肝細胞がん米国人105例の全エクソン解読(用語説明3)データを合わせた合計608例のゲノムデータを用いて、発がん要因や人種間における突然変異のパターンの相違点について解析を行いました。肝細胞がんゲノムにおける12種類の体細胞突然変異を6種類にまとめ(DNAはA:T、C:Gの相補的塩基からなる2本鎖であるため、例えばC>AとG>Tを区別できない)、さらに体細胞突然変異の周囲の配列情報として直前と直後の塩基(各4種類)を加え、合計で6x4x4=96種類に分類してデータとして用い、臨床背景との相関について解析し、さらに新しい解析技術の非負値行列因子分解(Non-negative matrix factorization:用語説明6)の手法を用いて、どのような体細胞突然変異パターンの組み合わせがどの程度貢献しているのかについて解析を行いました。
肝炎ウイルス以外の人種に特徴的な発がん要因の存在を推定
突然変異の分類は肝炎ウイルスの種類(B型肝炎、C型肝炎、非ウイルス性)とは関連しない一方で(図3右)、今回のデータから3種類の特徴的な変異パターン(シグネチャーA、シグネチャーB、シグネチャーC)を抽出することができ、それを人種別グループに分けて解析を行ったところ、シグネチャーAは日本人の肝細胞がんに特徴的な変異パターンであることが判明し、人種背景(日本人、中国人、ヨーロッパ人種)とは強い相関を示すことが分かりました(図3左、図4)。特徴的な変異パターンは、発がん要因と密接に関連するものであることからこの結果は、日本人に特徴的であり、かつ肝炎ウイルス感染ではない未知の発がん要因が存在していることを示唆しています。
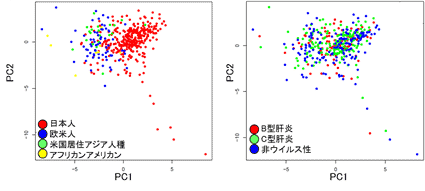
図3:変異データの主成分解析
左 人種背景ごとに分けた場合、統計値P= 2.2e-16
右 肝炎ウイルスの種類ごとに分けた場合、統計値P=0.35)
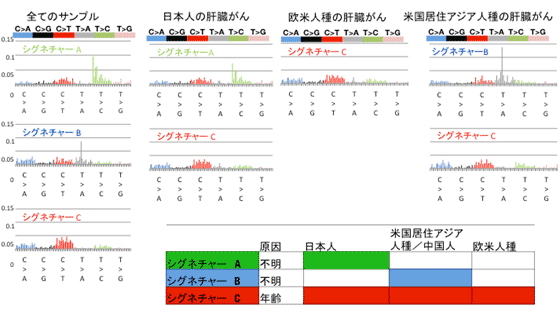
図4:非負値行列因子分解解析による変異パターンの抽出。全てのサンプルを用いた場合、3つのパターン(シグネチャーA、シグネチャーB、シグネチャーC)が抽出された。更に人種ごとの解析では、シグネチャーAは日本人の肝細胞がんでのみ、シグネチャーBは米国在住アジア人種の肝細胞がんでのみ、シグネチャーCは全ての人種で共通して検出された。
肝細胞がんにおける現状と今後の期待
肝細胞がんは、肝がんの90%を占めており、日本での肝がんの死亡数は第5位(2013年人口動態統計で、世界全体でも肺がんに続いて第2位を占めています(WHO Cancer Fact Sheet update 2014)(外部サイトにリンクします)。日本・韓国・中国といった東アジア並びにアフリカでの発生率が高いことが知られていますが、近年欧州や米国でも患者数が増加しており、国際的にもその克服が強く期待されている重要ながんです。 B型並びにC型肝炎ウイルス感染(日本ではC型肝炎陽性が70%を占める)が重要な危険因子として知られていますが、近年ではB型肝炎ワクチンの開発や肝炎治療成績の向上により、非肝炎ウイルス性(肥満、糖尿病、アルコール性肝炎等)の肝細胞がんが漸増しています。肝細胞がんの治療法としては、手術療法以外に経皮的ラジオ波焼灼療法、血管塞栓療法(用語説明7)などが行われていますが、分子標的抗がん剤ではソラフェニブしか認可されておらず、十分な治療法の開発が進んでいないのが現状です。
国立がん研究センター中央病院 肝胆膵内科長の奥坂拓志は、今回の研究成果について以下のように述べています。
「肝細胞がんにおけるB型、C型肝炎ウイルス以外の原因や人種差の解明に寄与しうる重要な研究と考えます。また、この研究をきっかけに、治療薬の少ない本疾患においても新しい有効な薬剤開発が推進することが期待されます。」
がんゲノム解析の現状と国際がんゲノムコンソーシアム
がんは正常な遺伝子(ゲノム)にさまざまな原因によって誘導される傷(ゲノム異常)が蓄積した結果として発生することが知られています。従って、がんで起こっているゲノム異常を解明することで、どの遺伝子に傷が入ったことでがんになったのかを知ることができます。いわゆる次世代シークエンサー(用語説明2)と呼ばれる新たな解析技術が開発され、ヒトゲノムを丸ごと解読することが可能となり、がん細胞のゲノムについてもその異常全体像の解明が急速に進んでいます。さらに、こうしたがん細胞に特異的なゲノムの傷を標的とした抗がん剤(いわゆる分子標的薬)が副作用の少ない有効な治療法として開発が進み、がんゲノム解読の結果を活用した新たな治療法の開発が迅速に進められるようになってきています。しかし一方で、多くのがんでは全体の10%以下の症例でしか見られない低頻度な異常がたくさん見られることが明らかとなり、それぞれのがんのゲノム異常全体を理解するためには数百例といった大規模な解析が必要であることも分かってきています。
こうした背景の下、世界各国を通じて臨床的に重要ながんを選定し、国際協力でそれらのがんについて少なくとも500例の解析を行い、そのゲノム変異の姿を明らかにするための国際共同プロジェクトとして、2008年に国際がんゲノムコンソーシアム(ICGC(外部サイトにリンクします))が発足しました。現在アジア、オーストラリア、ヨーロッパ、南北アメリカの16か国およびEUの組織が参画して、74個のがん腫についての大規模ゲノム研究プロジェクトが精力的に遂行されています。日本からは、国立がん研究センター・東京大学・理化学研究所が共同で肝細胞がんを対象としたプロジェクトを進めています。また米国でもTCGA(がんゲノムアトラスプロジェクト:The Cancer Genome Atlas、以下TCGA)プロジェクトとして、肝細胞がんを含めた20種類についてがんゲノム解読プロジェクトが進行中です。
遺伝情報を子孫に伝えるゲノムを形作る核酸(DNA)にはT,C,G,Aという塩基が含まれており、その塩基の種類により4種類に分類されます。がん細胞で見られる突然変異とはこの4種類の塩基が別の塩基に置き換わり核酸の情報が変化することであり、その種類は4x3=12通りとなります。ICGCプロジェクトでは現在までに10,000例を超えるさまざまながんの変異情報をデータベースで公開しており、そうしたがんゲノムビッグデータを用いた解析結果から、こうした突然変異の起こり方ががんの発生原因(発がん因子)と密接に関連していることが解明されてきています。例えば、たばこに含まれる発がん物質によって引き起こされる体細胞突然変異はCがAに(C>Aと表記)、あるいはGがT(G>T)に変化する2種類が大部分を占めています。更に全く新しい研究として、ゲノム解読ビッグデータを解析することで、色々ながんの原因を演繹的に探索することも可能となっています。
発表雑誌
雑誌名
Nature Genetics
論文タイトル
Trans-ancestry mutational landscape of hepatocellular carcinoma genomes
著者
Yasushi Totoki, Kenji Tatsuno, Kyle R. Covington, Hiroki Ueda, Chad J. Creighton, Mamoru Kato, Shingo Tsuji, Lawrence A. Donehower, Betty L. Slagle, Hiromi Nakamura, Shogo Yamamoto, Eve Shinbrot, Natsuko Hama, Megan Lehmkuhl, Fumie Hosoda, Yasuhito Arai, Kim Walker, Mahmoud Dahdouli, Kengo Gotoh, Genta Nagae, Marie-Claude Gingras, Donna M. Muzny, Hidenori Ojima, Kazuaki Shimada, Yutaka Midorikawa, John A. Goss, Ronald Cotton, Akimasa Hayashi, Junji Shibahara, Shumpei Ishikawa, Jacfranz Guiteau, Mariko Tanaka, Tomoko Urushidate, Shoko Ohashi, Naoko Okada, Harsha Doddapaneni, Min Wang, Yiming Zhu, Huyen Dinh, Takuji Okusaka, Norihiro Kokudo, Tomoo Kosuge, Tadatoshi Takayama, Masashi Fukayama, Richard A. Gibbs, David A. Wheeler(責任者),Hiroyuki Aburatani(責任者),Tatsuhiro Shibata(責任者)DOI番号:doi:10.1038/ng.3126
用語説明
ドライバー遺伝子
がん遺伝子・がん抑制遺伝子といった、がんの発生・進展において直接的に重要な役割を果たす遺伝子をドライバー遺伝子と呼ぶ。がんの発生過程においては、ゲノム変異が起こりやすい状態(いわゆるゲノム不安定性)となるため、がんの発生には無関係な遺伝子にもランダムに変異が起こることが知られている(背景変異、あるいはパッセンジャー遺伝子と呼ばれる)。従って、統計的解析によって、本物の異常(ドライバー遺伝子)と背景異常(パッセンジャー遺伝子)を区別する必要がある。ドライバー遺伝子は低分子阻害剤や抗体医薬などさまざまな分子治療の標的として有望である。
高速シークエンサー(次世代シーケンサー)
2003年のヒトゲノム計画終了後、ヒトゲノムの配列30億塩基対を1,000ドル以下のコストで解読すべく、欧米の政府や企業は技術開発を行ってきた。通常のサンガーシーケンス法と比べて、超大量のDNAシーケンス反応を並列して行う技術であり、最新の第2世代解析機器の場合、最大6日間で約1兆個の塩基配列を解読することができる(ヒトゲノム10人分に相当する)。
全エクソン解読と全ゲノム解読
ヒトの場合、実際にタンパク質の遺伝情報をコードしている領域(エクソンと呼ばれる)はゲノム全体の1から2%に過ぎない。従ってその領域に着目して解析を行なうことで、細胞の中で機能しているタンパク質の異常を包括的かつ効率的に同定することができる。この手法は全エクソン解読と呼ばれる。具体的には特殊な手法により、ゲノム全体からエクソン領域のみをあらかじめ濃縮し、高速シークエンサーにて解読を行なう。一方、エクソンを含めたゲノム全体の解読を行なう場合は、全ゲノム解読と呼ばれる。
テロメラーゼ
酵母からヒトまで真核生物の染色体末端にはテロメアという構造があり、染色体の安定性において重要である。しかし、細胞分裂の度にテロメアは短縮していくことから、細胞老化や不死化といった現象と深い関連があり、テロメアを維持するための酵素としてテロメラーゼが発見された(発見者は2009年にノーベル生理学・医学賞を受賞)。ヒトの正常な細胞ではテロメラーゼの発現は幹細胞、生殖細胞など限られた細胞にしか認められないが、無限増殖を行なうがん細胞においては、細胞老化を回避し、不死化を獲得するために異常なテロメラーゼの発現増加が起こることが知られている。
PIK3CA/mTOR経路
増殖因子による細胞増殖刺激情報伝達を担う分子経路の一つ。がん細胞において、この情報伝達経路に属する分子に突然変異等の異常が起こり、この経路の恒常的な活性化、すなわち細胞増殖のアクセルが入ったままの状態、が起こっていることが知られている。重要な創薬標的の一つであり、すでにこの経路を抑制するような分子標的薬(PIK3CA阻害剤やmTOR阻害剤)の開発が進められている。
Non-negative matrix factorization:非負値行列因子分解
0か正の値(非負値)のみからなるデータ(行列)を解析する手法。非負値行列を2つの非負値行列に分解するシンプルな解析アルゴリズムであるが、音響信号、画像データ、文書データの解析といったさまざまな分野への応用が可能である。例えば顔画像からの各パーツの特徴抽出等で使用され、その有効性が注目されている。がんにおける体細胞変異における特徴的なパターン抽出という目的においても有用であることが今回の検討でも示された。
問い合わせ先
内容に関するお問い合わせ
- 国立がん研究センター 研究所 がんゲノミクス研究分野 分野長
(東京大学医科学研究所附属ヒトゲノム解析センター ゲノムシークエンス解析分野 教授)
柴田 龍弘(しばた たつひろ)
電話番号:03-3542-2511(内線3123)
ファクス番号:03-3547-5137
Eメール:tashibat●ncc.go.jp(●を@に置き換えてください) - 東京大学 先端科学技術研究センター ゲノムサイエンス 教授
油谷 浩幸(あぶらたに ひろゆき)
電話番号:03-5452-5352
ファクス番号:03-5452-5355
Eメール:haburata-tky●umin.ac.jp(●を@に置き換えてください)
その他のお問い合わせ
- 独立行政法人国立がん研究センター 広報企画室
電話番号:03-3542-2511(代表)
ファクス番号:03-3542-2545
Eメール:ncc-admin●ncc.go.jp(●を@に置き換えてください)
関連ファイル
PDFファイルをご覧いただくには、Adobe Readerが必要です。Adobe Readerをお持ちでない方は、バナーのリンク先から無料ダウンロードしてください。