肺がんの新たな治療戦略へ期待
~免疫療法の治療効果の改善へ~
2020年2月4日
名古屋大学
国立がん研究センター
名古屋大学大学院医学系研究科分子細胞免疫学の 西川 博嘉 教授(国立がん研究センター研究所腫瘍免疫研究分野分野長、先端医療開発センター免疫TR分野分野長併任)らのグループは、肺腺がんの約半数に認められるepidermal growth factor receptor (EGFR)遺伝子変異(注1)が、がん細胞を殺傷する細胞傷害性T細胞(注2)や免疫応答を抑える働きをする制御性T細胞(注3)の移動をコントロールすることで、がん免疫療法(注4)に抵抗していることを明らかにしました。
現在、肺がんに対する抗PD-1抗体(注5)等の免疫療法の有効性が示されていますが、肺がんの中でも肺腺がんの約半数で認められるEGFR遺伝子変異陽性例では、がん免疫療法が効きにくいことが報告されています。その原因の一つとして、体細胞変異(注6)の数が少ないことが挙げられています。これは遺伝子変異により生じる異常たんぱく質(異物)が少なく、異物を除去するための免疫応答が起こりにくいがんのタイプであると推察されています。本研究ではさらなる詳細な解析により、EGFR遺伝子変異陽性例では、がん組織の中に細胞傷害性T細胞の入り込んでいく数が少なく、一方で、制御性T細胞が多いことを明らかにしました。なぜ制御性T細胞が多いのかを検討をし、EGFR遺伝子変異陽性の肺がんが、制御性T細胞を呼び寄せる化学物質[ケモカイン(注7)(CCL22)]を多く産生する一方で、がん細胞を殺傷する細胞傷害性T細胞を呼び寄せる化学物質[ケモカイン(CXCL10やCCL5)]の産生が少ないことを解明しました。そこで、がんを移植したマウスを用いて、EGFRシグナル(注8)を阻害した状態で抗PD-1抗体を用いると、肺がんの治療効果が改善することが明らかになりました。これらの研究成果は、今後のがん免疫療法の治療効果の改善につながる可能性があります。
本研究成果は、2020年2月1日付け(日本時間午前4時)米国科学雑誌「Science Immunology」電子版に掲載されました。
ポイント
- 肺がんに対する抗PD-1抗体等の免疫療法の有効性が示されていますが、肺がんの中でも肺腺がんで認められるepidermal growth factor receptor (EGFR)遺伝子変異陽性例では、がん免疫療法が効きにくいことが報告されていますが、その原因については十分に解明されていません。
- 本研究では、EGFR遺伝子変異陽性肺がん細胞が制御性T細胞を呼び寄せ、一方、がん細胞傷害性T細胞を遠ざけることで、がん免疫療法に抵抗していることを明らかにしました。
- 以上の免疫抑制メカニズムの解明に基づき、EGFR遺伝子シグナル阻害剤とがん免疫療法の併用により、より良好な治療効果が得られる可能性を示しました。
1.背景
非小細胞肺がんの大部分を占める肺腺がんでは、約半数程度でepidermal growth factor receptor (EGFR)遺伝子変異が認められます。EGFR遺伝子変異は、EGFRシグナルを恒常的に活性化させることで発がんに寄与するドライバー遺伝子変異であり、EGFRチロシンキナーゼ阻害剤(注9)を用いるとがんに対する劇的な縮小効果が得られ、実臨床で広く使用されています。しかしながら、やがてEGFRチロシンキナーゼ阻害剤に抵抗性の肺がん細胞が出現することで治療に抵抗性になることが問題となっていました。
一方で、近年、肺がんに対する新たな治療法として、抗PD-1抗体等を用いたがん免疫療法の有効性が示され、注目されていますが、EGFR遺伝子変異陽性肺がん(以下、EGFR陽性肺がん)に対しては治療効果が乏しいことが報告されていたことから、本研究ではその免疫学的メカニズムを解明し、新たな治療法の開発につながる基礎検討を行いました。
2.研究成果
EGFR陽性肺がんでは、体細胞変異数が少ないことが免疫療法の効果が乏しい要因の一つと言われていることから、実際の肺がん症例から採取したがん細胞を用いて遺伝子解析を行い、体細胞変異数について検討しました。EGFR陽性肺がんでは、EGFR陰性肺がんと比較し、体細胞変異が明らかに少ないことが示されました(図1)。これは遺伝子変異により生じる異常たんぱく質(異物)が少ないことを意味し、異物を除去するための免疫応答が起こりにくいがんのタイプであることが示唆されました。
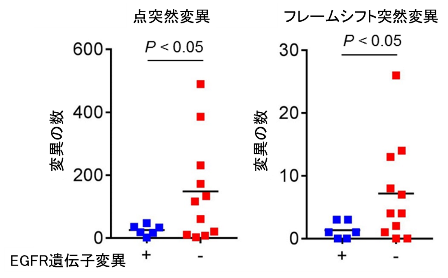
図1. EGFR遺伝子変異の有無による肺がんの体細胞変異数の比較検討
肺がん患者から採取した肺がん細胞を用いて遺伝子解析(Exome解析)を行い、体細胞変異数(点突然変異およびフレームシフト変異)をEGFR遺伝子変異の有無で比較検討しました。
次に、肺がん患者のがん組織にどのような免疫細胞が浸潤しているか解析しました。免疫染色を用いてがん細胞を殺傷する傷害性T細胞および免疫抑制に働く制御性T細胞の数を評価すると、EGFR陽性肺がんでは、EGFR陰性肺がんと比較し、細胞傷害性T細胞が少なく、一方で、制御性T細胞が多浸潤していることが明らかとなりました(図2)。
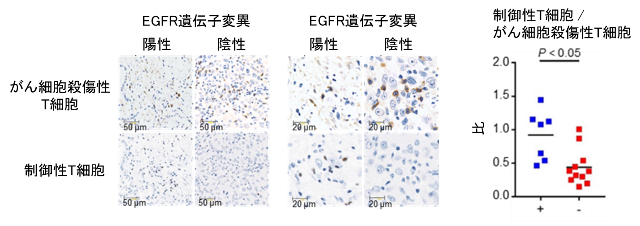
図2. EGFR遺伝子変異の有無による肺がん組織内の免疫細胞数の免疫染色を用いた比較検討
肺がん患者から採取した肺がん組織を用いて免疫染色を行い、がん組織内に浸潤している細胞傷害性T細胞と制御性T細胞の数をEGFR遺伝子変異の有無で比較検討しました。
がん組織から抽出したリンパ球の割合を、細胞を1個ずつ定量測定する機器であるフローサイトメトリーおよびCyTOF(マスサイトメトリー)という測定方法用いて解析すると、免疫染色の結果と同様に、EGFR陽性肺がんではがん細胞を殺傷する細胞傷害性T細胞の割合が低く、一方で、制御性T細胞が高頻度に集まっていることが示されました(図3)。
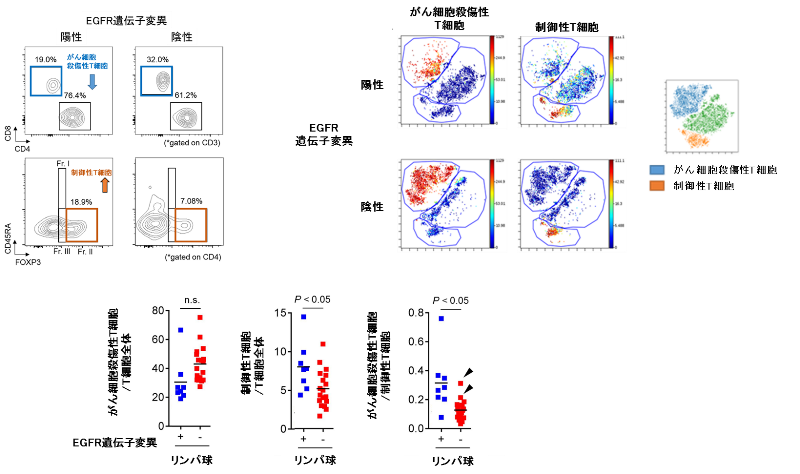
図3. EGFR遺伝子変異の有無のよる肺がん組織内の免疫細胞数のフローサイトメトリーおよびCyTOFを用いた比較検討
肺がん患者から採取した肺がん組織内から抽出した免疫細胞(リンパ球)を、抗体を用いて染色し、フローサイトメトリーやCyTOF(マスサイトメトリー)を用いて、がん細胞殺傷性T細胞や制御性T細胞の数や割合をEGFR遺伝子変異の有無で比較検討しました。
以上より、EGFR陽性肺がんは、特徴的な免疫抑制性の腫瘍環境(がん細胞を殺傷する細胞傷害性T細胞の浸潤が少なく、制御性T細胞の浸潤が多い)であることが示唆されました。
なぜ、このような特徴的な免疫抑制性の腫瘍環境となっているのかを解明するため、肺がん細胞株を用いて解析を行いました。EGFR陽性の肺がん細胞では、免疫細胞を呼び寄せる作用のある化学物質(ケモカイン)の分泌能が重要な働きをしていることが明らかとなりました。EGFR陽性肺がんでは、EGFRシグナルが強く、制御性T細胞を呼び寄せる化学物質(ケモカインの一つであるCCL22)の分泌能が高いことが示されました、一方で、EGFRチロシンキナーゼ阻害剤を用いてシグナルを抑制すると分泌能が低下することが確認されました(図4)。
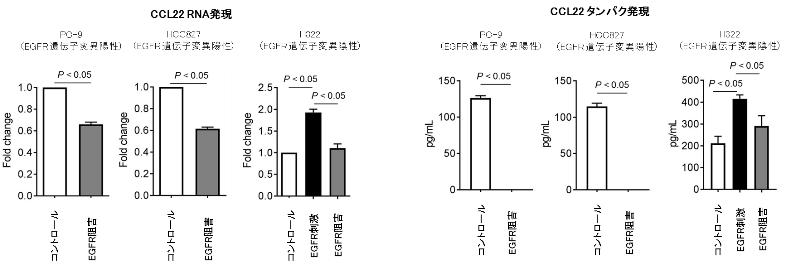
図4. 肺がん細胞株を用いたEGFRシグナル刺激 / 阻害によるCCL22発現の検討
肺がんの細胞株(EGFR遺伝子変異陽性株 2種類、陰性株1種類)を用いて、EGFRシグナル阻害/刺激によりCCL22の分泌能の変化を検討しました。
また、がん細胞を殺傷する細胞傷害性T細胞を誘導するケモカインであるCXCL10やCCL5(CCL5については図表を割愛)については、EGFRシグナルが活性化すると分泌能が低下し、EGFRシグナルが阻害されると上昇することが確認されました(図5)。
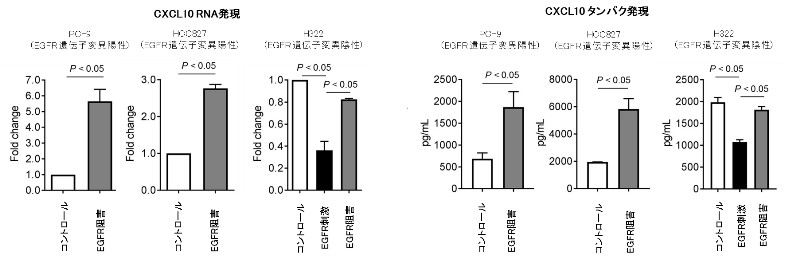
図5. 肺がん細胞株を用いたEGFRシグナル刺激 / 阻害によるCXCL10発現の検討
肺がんの細胞株(EGFR遺伝子変異陽性株 2種類、陰性株1種類)を用いて、EGFR阻害/刺激によりCXCL10の分泌能の変化を検討しました。
以上より、これらのケモカインの分泌の変化がEGFR陽性肺がんの特徴的な免疫抑制性環境の形成に寄与している可能性が示唆されました。
これらのケモカイン発現のメカニズムを解明するため、それぞれのケモカインの合成を調節している転写因子(注10)を探索したところ、CCL22はJUNという転写因子が、CXCL10 / CCL5についてはIRF1という転写因子が、それぞれのケモカインの分泌を調節していることが明らかになりました。JUNやIRF1をノックダウンする(si-JUNやsi-IRF1といった分子を用いてJUNやIRF1の発現を落とす)ことで、それぞれCXCL10とCCL22の発現が低下していることが確認されました。これらの結果より、JUNがCCL22の発現を、IRF1がCXCL10の発現を制御していることが示されました。これらの転写因子はEGFRシグナルの活性化によりJUNは上昇、IRF1は低下することが確認されました。
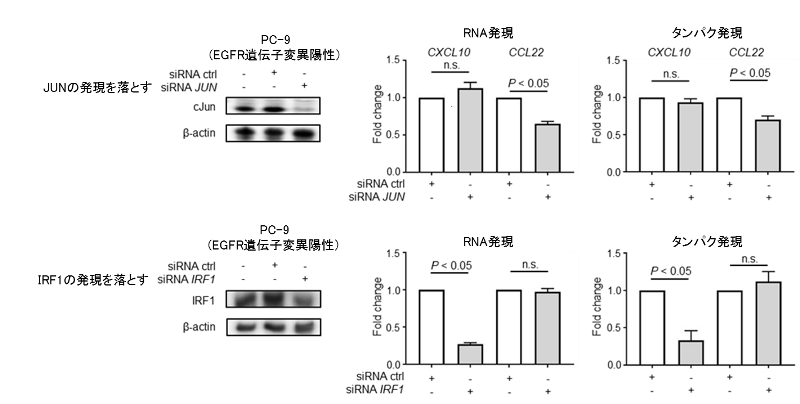
図6. 肺がん細胞株を用いたJUN / IRF1ノックアウト時のCXCL10 / CCL22の発現変化の検討
肺がん細胞株(EGFR遺伝子変異陽性株)を用い、JUNやIRF1の発現をノックアウト(si-JUNやsi-IRF1といった分子を用いてJUNやIRF1の発現を落とす)し、CXCL10やCCL22の発現の変化を検討しました。
以上の結果を踏まえ、EGFRシグナルを阻害した状態で抗PD-1抗体といったがん免疫療法を実施すると抗腫瘍効果が高まる可能性を検証するため、EGFR遺伝子変異を挿入したマウス細胞株MC-38をC57BL/6Jマウス(注11)に移植し、EGFRチロシンキナーゼ阻害剤、抗PD-1抗体、その両方を投与してみると、併用群で最も強く腫瘍増殖が抑制され、生存期間の延長を認められました(図7)。
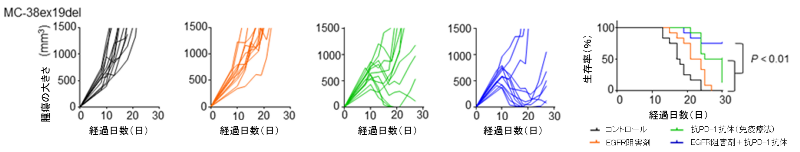
図7. EGFR遺伝子変異陽性肺がん細胞株を移植したマウスを用いた、EGFR阻害剤と抗PD-1抗体の併用効果の検討
EGFR遺伝子変異陽性肺がん細胞株を移植したマウスを用い、コントロール、EGFRチロシンキナーゼ阻害剤、抗PD-1抗体(がん免疫療法)、EGFRチロシンキナーゼ阻害剤と抗PD-1抗体の両方を投与し、4群で腫瘍の大きさやマウスの生存率を検討しました。
3.今後の展開
EGFR陽性肺がんにがん免疫療法が治療効果を発揮しにくい原因として、体細胞変異数が少ないことが考えられてきました。本研究により特徴的な免疫抑制性の腫瘍環境が構築されていることが明らかになりました。EGFR陽性肺がんではEGFRシグナルは、従来考えられてきたようにがん細胞の増殖に関わるのみならず、ケモカイン分泌を介して免疫抑制性の環境を作り上げていることが示されました。これは、従来の発がんを誘発する遺伝子変異であるドライバー遺伝子が細胞増殖に関わるという概念を超えた新しい概念と考えられます。このような免疫抑制性の腫瘍環境を打破するには、EGFRシグナル活性を阻害した上で、がん免疫療法を行うと有効である可能性が示唆され、今後の肺がんの新たな治療戦略につながる可能性が示唆されます。
4.用語説明
(注1) EGFR遺伝子変異
EGFRは、がん細胞が増殖をするためのスイッチのような役割を果たしているタンパク質で、がん細胞の表面にたくさん発現しています。このEGFRを構成する遺伝子の一部に遺伝子変異があると、細胞を増殖させるスイッチが常にオンとなっているような状態となり、細胞が限りなく増殖してしまい、がんを引き起こします。このような発がんを誘発する遺伝子変異のことを、ドライバー遺伝子変異と呼びます。
(注2) 細胞傷害性T細胞
体内の免疫細胞のうち、CD8分子陽性のT細胞集団で、標的抗原を発現する細胞を殺傷します。がん免疫では、がん細胞に発現するがん抗原を認識してがん細胞を殺傷します。
(注3) 制御性T細胞
体内の免疫細胞のうち、CD4分子陽性かつFoxP3分子陽性のT細胞集団で、免疫抑制機能を発揮します。体内で起こる様々な免疫応答を抑制する働きがありますが、がん免疫では抗腫瘍免疫応答を抑制してしまいます。
(注4) がん免疫療法
生体内の免疫細胞ががん細胞を異物として認識し、攻撃することを利用したがん治療法です。従来の標準治療法として知られている化学療法・外科的治療・放射線療法よりも副作用が少なく長期的な治療効果が期待できることから、近年大きな注目を集めています。
(注5) 抗PD-1抗体
Programmed cell Death 1 (PD-1)に対する抗体です。ヒトではニボルマブ(Nivolumab)やペンブロリズマブ(Pembrolizumab)として医薬品承認を受けています。PD-1はがん細胞のPD-L1やPD-L2分子と相互作用することで細胞傷害性T細胞の活性化を阻害します。抗PD-1抗体はこの抑制性の相互作用をブロックし、細胞傷害性T細胞の活性化を維持します
(注6) 体細胞変異
遺伝的なDNAの変異とは異なり、分化や生育の過程で後天的に獲得したDNA変異のことを指します。がんの原因となる体細胞変異(ドライバー遺伝子変異)を特定することもできます。その一つがEGFR遺伝子変異です。
(注7) ケモカイン
リンパ球を引き寄せる化学メディエーターです。細胞傷害性T細胞や制御性T細胞といったリンパ球を局所に引き寄せ、炎症の形成、抑制に関与するサイトカインの一種です。
(注8) EGFRシグナル
EGFRとその下流に存在する様々な分子を含めた総称であり、EGFRが活性化することで下流に存在する分子Aが活性化され、分子Aが分子Bを、分子Bが分子Cをといったように、下流に向かって順番に活性化されることで、細胞増殖、分化などを誘導する経路のことである。
(注9) EGFRチロシンキナーゼ阻害剤
EGFRの活性化に一番重要な部分であるチロシンキナーゼ部を選択的に阻害することで、EGFRシグナルの活性化を落とし、がん細胞の増殖を抑制する薬です。ヒトでは、ゲフィチニブ(Gefitinib)、エルロチニブ(Erlotinib)、アファチニブ(Afatinib)、オシメルチニブ(Osimertinib)が医薬承認を受けています。
(注10) 転写因子
遺伝子からタンパク質への翻訳・変換を手助けするタンパク質の総称です。
(注11) C57BL/6Jマウス
ヒトの疾患のモデルとして用いられる遺伝子改変マウスとして、最も広く使用されている種類のマウスを指します。
5.発表雑誌
掲雑誌名
Science Immunology (米国時間1月31日14時付の電子版)
論文タイトル
Blockade of EGFR improves responsiveness to PD1 blockade in EGFR-mutated non-small cell lung cancer
著者
Eri Sugiyama,1,2 Yosuke Togashi,2 Yoshiko Takeuchi,2 Sayoko Shinya,2 Yasuko Tada,2 Keisuke Kataoka,3 Kenta Tane,4 Eiichi Sato,5 Genichiro Ishii,6 Koichi Goto,7 Yasushi Shintani,8 Meinoshin Okumura,8 Masahiro Tsuboi,4 and Hiroyoshi Nishikawa,1,2*
所属
1. Department of Immunology, Nagoya University Graduate School of Medicine, Nagoya 466–8550, Japan.
2. Division of Cancer Immunology, Research Institute/Exploratory Oncology Research & Clinical Trial Center (EPOC), National Cancer Center, Tokyo 104-0045/Chiba 277-8577, Japan.
3. Division of Molecular Oncology, Research Institute, National Cancer Center, Tokyo 104-0045, Japan.
5. Department of Pathology, Institute of Medical Science, Tokyo Medical University, 160-0023 Tokyo, Japan.
4. Division of Thoracic Surgery, 6. Division of Pathology, 7. Division of Thoracic Oncology, National Cancer Center Hospital East, Chiba 277-8577, Japan.
8. Department of General Thoracic Surgery, Osaka University Graduate School of Medicine, Osaka 565-0871, Japan.
DOI
10.1126/sciimmunol.aav3937
6.広報担当
名古屋大学医学部・医学系研究科総務課総務係
電話番号:052-744-2228 FAX:052-744-2785
E-mail:iga-sous●adm.nagoya-u.ac.jp(●を@に置き換えてください)
国立がん研究センター 企画戦略局 広報企画室(柏キャンパス)
電話番号:04-7133-1111(代表) FAX:04-7130-0195
E-mail:ncc-admin●ncc.go.jp(●を@に置き換えてください)
関連ファイル
PDFファイルをご覧いただくには、Adobe Readerが必要です。Adobe Readerをお持ちでない方は、バナーのリンク先から無料ダウンロードしてください。