大規模公共トランスクリプトームデータを活用した疾患関連変異の新規スクリーニング手法の開発蓄積が進むオミクスデータからの自律的な知識獲得基盤の実装に向けて
2022年9月29日
国立研究開発法人国立がん研究センター
発表のポイント
- データの特性を考慮した新しいアルゴリズムを開発し、トランスクリプトームデータ(注1)からスプライシング異常(注2)による疾患関連変異を検出する新しい情報解析手法である IRAVNetを開発しました。
- Amazon Web Servicesのクラウド(注3)上や国内研究機関スパコン(注4)上に大規模シークエンス解析プラットフォームを構築、20万件を超える公共トランスクリプトームデータにIRAVNetを適用し、約3,000の新規疾患関連変異のカタログを得ました。
- 今後も蓄積が進むオミクスデータ(注5)に対して本研究で開発した情報解析基盤を適用することで、さらに多くの新規疾患関連変異を自律的に同定する仕組みを構築することが期待できます。
概要
国立研究開発法人国立がん研究センター(理事長:中釜 斉、東京都中央区) 研究所(所長:間野 博行)のゲノム解析基盤開発分野 白石 友一分野長は、がんRNA研究分野 吉見 昭秀分野長らとの共同研究により、公共データレポジトリ(注6)に登録されている数十万検体を超える規模のトランスクリプトームデータを活用して、疾患に関連するゲノム変異を探索する新しい情報解析基盤の開発に成功しました。
本研究では、ゲノム変異によりスプライシング異常、特にイントロン残存(注7)が生じる場合にトランスクリプトームデータに変異が観測されるという特性を利用して、トランスクリプトームデータからイントロン残存を引き起こす変異をスクリーニングする方法(IRAVNet)を開発しました。さらに、開発した方法論を大規模データに適用するために、Amazon Web Servicesのクラウド上の計算環境や国内研究機関におけるスーパーコンピューター上で動作する情報解析基盤を構築しました。Sequence Read Archive(注8)に登録されている20万件を超えるトランスクリプトームデータに適用し、がんドライバー遺伝子、難病に関連する遺伝子など、約3,000の新規疾患関連変異の候補を検出し、カタログ化しました。本研究で構成した変異カタログにより、今後のゲノム医療実装における治療標的の同定、疾患診断の精緻化に繋がることが予想できます。また、今後加速度的に蓄積が進むオミクスデータに本研究で開発した情報解析基盤を適用することで、さらに多くの新規疾患関連変異を自律的に同定する仕組みを構築することが期待できます。
本研究成果は、「Nature Communications」に2022年9月29日掲載されました。
背景
ゲノム解析の有効性が広く検証され、現在、世界的に国家規模のゲノムプロジェクトが進められており、ゲノム解析は医療システムに大きな変革をもたらすことが期待されています。その中で患者のゲノムシークエンスによって得られる膨大な変異の中から疾患に関連する変異を同定することは、ますます重要な課題となっております。
疾患に大きな影響を及ぼす変異の最も重要なクラスの1つは、スプライシングに異常を引き起こす変異であり、ヒトの疾患関連変異の15%から60%を占めると言われています。一方でスプライシングのメカニズムはまだ未解明なところが多く、スプライシング異常を引き起こすゲノム変異の予測、またデータベース化は十分に進んでおりません。これまでのスプライシング変異の同定を目指した多くの研究では、ゲノムデータとトランスクリプトームデータの両方を使って、スプライシング異常とゲノム変異の有無の相関を見るアプローチが一般的でした。しかしながら、このアプローチのためにはゲノムとトランスクリプトームの両方のデータが提供されているデータセットが必要であり、こうした状況はさほど一般的ではありませんでした。その一方で、Sequence Read Archive などの公共データレポジトリには研究者が自由にアクセスできる数十万件規模のトランスクリプトームデータが配置されており、さらにデータの蓄積が加速度的に続いています。
研究方法
我々は、膨大な公共トランスクリプトームデータを最大限に活用するために、トランスクリプトームシーケンスデータのみを用いて、スプライシング異常の一形態であるイントロン残存を引き起こすゲノム変異を同定できる新規のアルゴリズム、IRAVNet (https://github.com/friend1ws/iravnet(外部サイトにリンクします)) を開発しました。この方法論は、ゲノム変異によりイントロン残存が生じた際に、トランスクリプトームシークエンスデータに該当のゲノム変異が観測されるという特性に着目して開発されました(図1)。
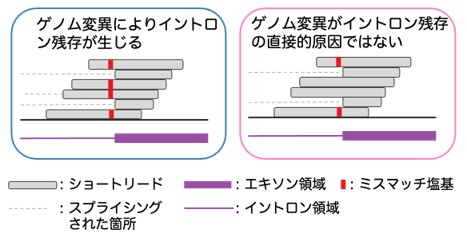
図1 イントロン残存変異周辺のRNA-seqの例
変異が、IRの原因となっている場合は、IRを示しているリード上に特異的に変異が観測できます。
(左)ゲノム変異がイントロン残存の直接的な原因となっている場合は、イントロン残存を引き起こしているリードに特異的にミスマッチ塩基が観測されます。
(右)イントロン残存の原因がゲノム変異ではない場合は、イントロン残存をサポートしているリードにミスマッチ塩基が観測されないこともあります。
さらに、SRAなどに登録されている大規模なトランスクリプトームに対してこの方法論を適用するために、我々はAmazon Web Servicesを利用したクラウドベース(図2)の解析プラットフォームと、国内研究機関(国立遺伝学研究所 生命情報・DDBJ センター、東京大学医科学研究所 ヒトゲノム解析センター)の計算クラスタを用いたプラットフォームの両方を開発しました。特にクラウド上の解析基盤においては、各解析ステップにおいて最適なインスタンスタイプの選択、適切なブロックストレージの確保、スポットインスタンスの利用など、利用コストを抑えるための様々な工夫がなされています。クラウドは自由度が高く、きめ細やかなプラットフォーム構築が可能であること、開発したプラットフォームを世界中で共有することが比較的容易にできるという利点があります。一方で、構築・運用にはクラウドの知識が不可欠であり、また利用料金に手間がかかるという問題もありました。国内研究機関スパコンは、アカデミアの研究者にとっては比較的安価で、料金体系がクラウドに比べてシンプルで、気軽に利用できるといった利点があります。
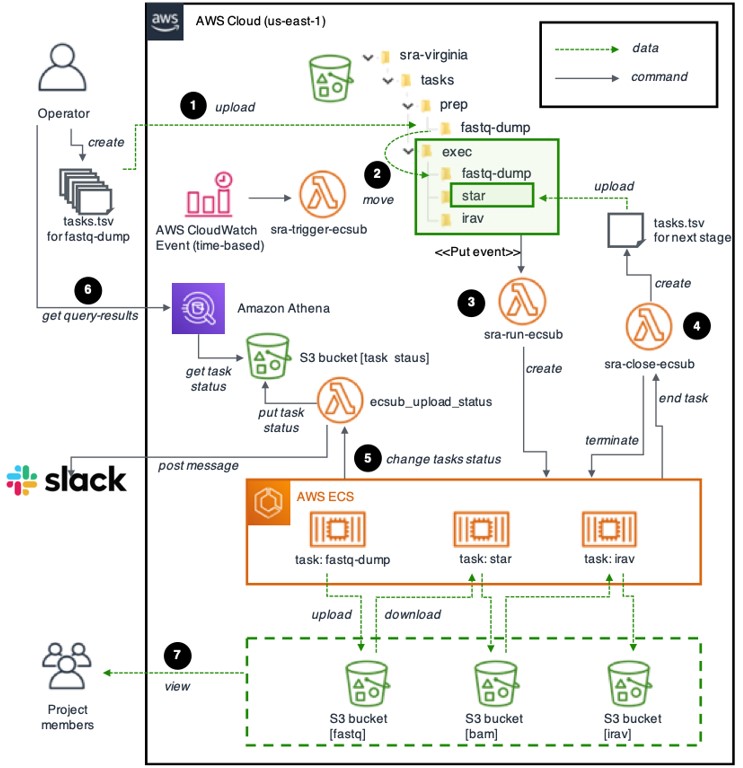
図2 Amazon Web Services上に構築した解析基盤
トランスクリプトームデータのダウンロード、アラインメント、IRAVNetの各種解析を自律的に実行することが可能です。
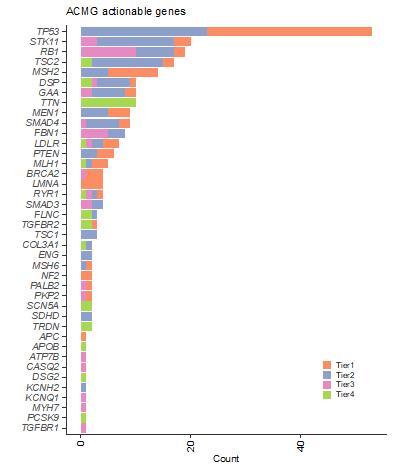
図3 本研究検出したACMG actionable geneにおける疾患関連変異の一覧
既知の疾患関連変異との位置関係によりTier分類がなされています。
Tier1はClinVar(注9)にスプライシング変異として登録されていた、「既知の」疾患関連変異、Tier2から4は「新規の」疾患関連変異の候補。
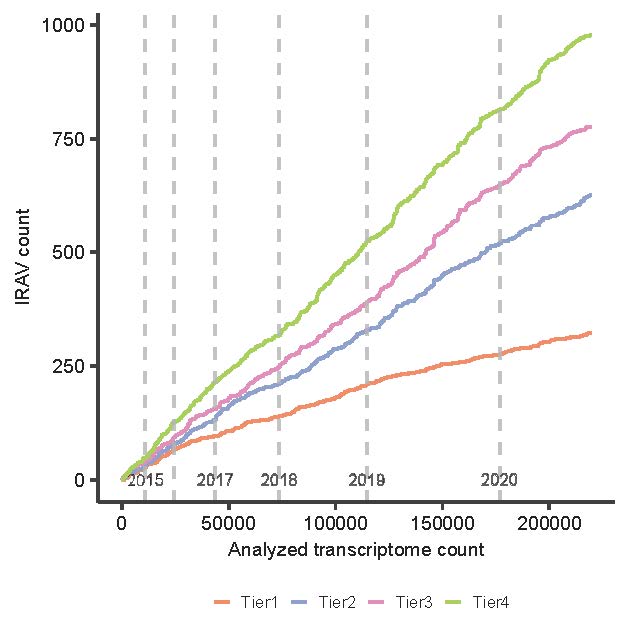
図4 解析データ数とそれに応じて検出される疾患関連変異の個数の関係性
データの登録された日付順の並べ替えがなされています。年度の間隔が大きくなり、データの蓄積が加速度的に進んでいることがわかります。
展望
今回の解析においては、イントロン残存変異を引き起こすゲノム変異に着目しましたが、現在はさらに別のタイプのスプライシング変異の同定を可能とする方法論の開発に着手しており、得られる疾患関連変異のクラスを広げることを目指して研究を進めています。本研究の一連の成果により、今後、がん・難病の全ゲノム解析プロジェクトが進む中で、意義不明変異の機能予測に役に立つことが期待されます。また、本研究で開発された解析プラットフォームは高度に自動化がなされており、今後のデータ登録に併せて実行する仕掛けを施すことで、自律的に病的変異を蓄積するシステムの構築も十分に可能であり、今後も増加を続けるオミクスデータの利活用方法に対して一石を投じる研究成果となりました。
発表論文
雑誌名
Nature Communications
タイトル
Systematic identification of intron retention associated variants from massive publicly available transcriptome sequencing data
著者
Yuichi Shiraishi†,*, Ai Okada†, Kenichi Chiba, Asuka Kawachi, Ikuko Omori, Raúl Nicolás Mateos, Naoko Iida, Hirofumi Yamauchi, Kenjiro Kosaki, Akihide Yoshimi
(†第一著者と同等の寄与, *責任著者)
掲載日
2022年9月29日
DOI
10.1038/s41467-022-32887-9
URL
https://www.nature.com/articles/s41467-022-32887-9(外部サイトにリンクします)
研究費
- 日本学術振興会
科学研究費補助金 基盤研究(B) 「大規模トランスクリプトームからの自律的知能獲得システム基盤の開発」 (代表: 白石 友一)
- 国立研究開発法人日本医療研究開発機構(AMED)
難治性疾患実用化研究事業 「長鎖・短差鎖シークエンス技術の統合による構造変異の検出と非翻訳領域情報を駆使した未診断症例の解決」 (代表: 小崎 健次郎)
用語解説
[注1]トランスクリプトームデータ
細胞中に存在している全てのメッセンジャーRNAの総体を表すデータ。本研究では、特に次世代シークエンサーによる配列情報の形で取得されたデータを利用している。
[注2]スプライシング異常
メッセンジャーRNAの前駆体は、「スプライシング」の過程において遺伝子配列から不要な部分であるイントロン領域が取り除かれ、成熟したメッセンジャーRNAとなる。このプロセスに異常が生じることをスプライシング異常という。この結果として、正常とは異なるタンパク質が生成されることや、タンパク質の発現量の低下が引き起こされる。
[注3] クラウド
ユーザーがインターネットを通じて、仮想的な計算機や周辺のアプリケーションを必要な時に必要な分だけ利用できるサービスの総称。特にAmazon Web Services, Google Cloud Platform, Microsoft Azureなどが有名である。
[注4] 国内研究機関スパコン
国立遺伝学研究所生命情報・DDBJ センターや東京大学医科学研究所ヒトゲノム解析センター等は国内の生命・医学研究者のために比較的安価に大規模計算環境を提供している。また、DDBJセンターのスパコンには、Sequence Read Archiveのデータがミラーリングされており、当該データの効率な利用が可能である。
[注5]オミクスデータ
ゲノム、トランスクリプトーム、プロテオーム、エピゲノム、メタボロームなどの網羅的な生体分子の情報を計測したデータのことを示している。
[注6]公共データレポジトリ
研究等で取得されたデータを、再利用可能な形で保存・管理し、インターネット上でオープンにアクセス可能な形で公開を行うウェブサイト・サーバーの総称。
[注7] イントロン残存
スプライシング異常の中でも、特にイントロン領域が適切に取り除かれずに残存してしまう現象のことである。
[注8] Sequence Read Archive
次世代シークエンサーで生成されたショートリードを格納しているデータレポジトリ。米国のNCBI (National Center for Biotechnology Information)、英国のEBI(European Bioinformatics Institute), 日本のDDBJ センター(DNA Data Bank of Japan)が共同で管理している。大部分のデータは、アクセス制限なしに、自由にダウンロードが可能である。
[注9]ClinVar
疾患に関連するゲノム変異の情報について、その信頼度や付加情報を含めて整備・収集しているデータベース。 https://www.ncbi.nlm.nih.gov/clinvar/(外部サイトにリンクします)
報道関係からのお問い合わせ先
研究に関する問い合わせ
国立研究開発法人国立がん研究センター
研究所 ゲノム解析基盤開発分野
担当者名:白石 友一
メールアドレス: yuishira●ncc.go.jp(●を@に置き換えください)
電話番号: 03-3542-2511(代表)
広報窓口
国立研究開発法人国立がん研究センター
企画戦略局 広報企画室
メールアドレス:ncc-admin●ncc.go.jp(●を@に置き換えください)
電話番号:03-3542-2511(代表)
関連ファイル
PDFファイルをご覧いただくには、Adobe Readerが必要です。Adobe Readerをお持ちでない方は、バナーのリンク先から無料ダウンロードしてください。