トップページ > 研究組織一覧 > 分野・独立ユニットグループ > 細胞情報学分野 > 研究プロジェクト > 研究プロジェクト(細胞情報学分野)
研究プロジェクト(細胞情報学分野)
発がん原因の解明
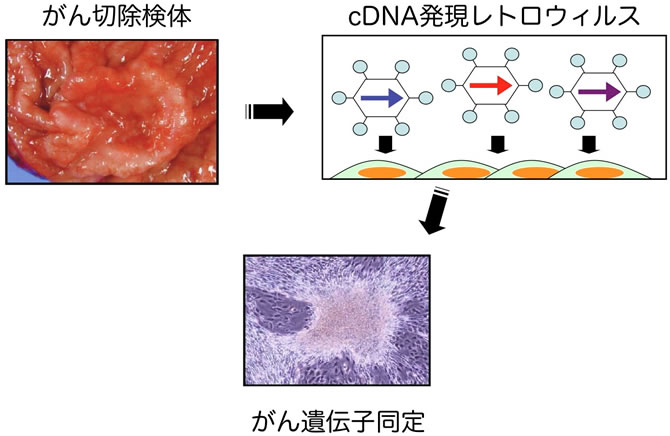
私たちは、発がんの直接的な原因遺伝子を発見する目的で、患者検体で発現している遺伝子の機能を効率よくスクリーニングするcDNA発現スクリーニングシステムを構築しました。本法を用いて肺がんの臨床検体からcDNA発現レトロウィルスライブラリーを構築し、3T3繊維芽細胞によるフォーカスフォーメーションアッセイを行ったところ、2~3週間で形質転換フォーカスが数十個生じたのです。これら異常細胞に挿入されたcDNAを増幅回収したところ、驚くべき事に、5’側は微小管会合タンパクの一種であるEML4(echinoderm microtubule-associated protein–like 4)のアミノ末端側約半分をコードし、3’側は受容体型チロシンキナーゼALK(anaplastic lymphoma kinase)の細胞内チロシンキナーゼドメインをコードするcDNAが同定されたのです(Nature 448:561)。
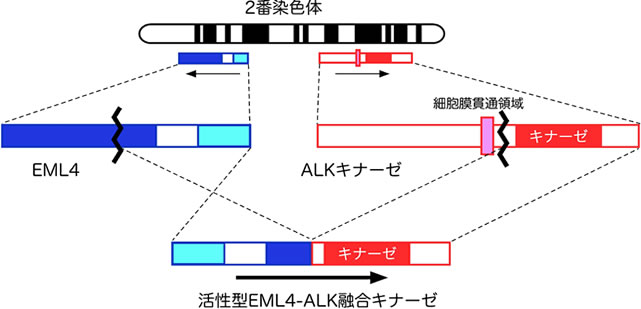
正常ALKは細胞膜に存在するチロシンキナーゼですが、2番染色体短腕内の微小な逆位の結果EML4と融合し、恒常的に活性化されて強力な発がん能を有する酵素となるのです。実際、私たちがEML4-ALKを肺胞上皮特異的に発現するトランスジェニックマウスを作成したところ、驚くべき事にそれらマウスは生後数週間のうちに両肺に数百個の肺腺がんを発症し、しかもこれらマウスに経口摂取可能なALK阻害剤を一日一度投与したところ一ヶ月以内に肺腺がんはほぼ消失しました。すなわちEML4-ALK陽性肺がんにおいては同融合遺伝子こそが発がんの主たる原因であり、だからこそその機能を抑制する薬剤は肺がんの全く新しい分子標的治療法となることが生体において証明されたのです(PNAS 105:19893)。
EML4、ALK両遺伝子は共に正常細胞内に存在しますが、両者が融合したEML4-ALK遺伝子は肺がん細胞内でしか存在しません。しかも両遺伝子は本来染色体内で反対向きに存在しているため、EML4-ALKの融合点を挟む様に設置したプライマーによるRT-PCR法は、同融合遺伝子が存在しない限り決してPCR産物を生じない高精度かつ高感度な肺がん診断法になると期待されます。実際10万個の正常細胞内に1個しか存在しないEML4-ALK陽性細胞を検出可能なことが確認されました(Clin Cancer Res 18:5682)。
EML4-ALKの発見を受けて数多くの製薬会社がALK阻害剤の開発に乗り出しましたが、最初に第I相臨床試験に入ったのはクリゾチニブでした。その成果は2010年のNew England Journal of Medicine誌に報告され、FISHを含む2種類の診断法で陽性になった症例において奏功率が8割を超え、完全寛解も生じるという目覚ましい治療効果が明らかになったのです(NEJM 363:1693)。またクリゾチニブは第I/II相試験の結果のみで2011年8月26日に米国FDAから薬剤として承認されました。2007年の私たちのEML4-ALKの発見からわずか4年での薬剤の承認・販売は、がんの治療薬開発史上最速のスピードと言えます。
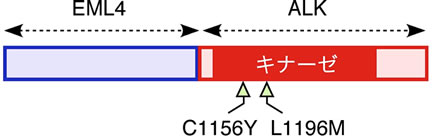
さらに私たちは、EML4-ALK陽性肺腺がん症例で当初はクリゾチニブが著効したものの、約半年後突然再発し薬剤耐性となった症例を経験しました。本症例の治療前と再発後の肺がん検体を次世代シークエンサーによって解析する事により、再発時にのみEML4-ALKのキナーゼドメイン内に新たな付加変異を2種類発見したのです(NEJM 363:1734)。これら変異の一つはALK内の1156番目のシステインをチロシンに置換し、もう一つは1196番目のイソロイシンをメチオニンに置換します。EML4-ALKを導入したBA/F3細胞はEML4-ALKの活性依存性に増殖しますが、培養上清にクリゾチニブを添加するとその濃度依存性に細胞死が誘導されます。ところがC1156YあるいはL1196Mを有するEML4-ALKはどちらもクリゾチニブ耐性になるのです。また既にこれら変異があっても有効な「第2世代のALK阻害剤」が次々と開発され、臨床試験に入っています。こうして私たちの発見により世界中の何万人もの肺がん患者さんの救命が実現しました。このように、直接的な発がん原因を見つけることで新たながんの特効薬がもたらされることが証明され、私たちはこの様なアプローチを更に展開しています。
次世代シークエンサーを用いたがんの網羅的ゲノム解析
次世代シークエンサーを用いることで300—600 Gbpもの塩基配列情報を一度の実験で得ることができるようになり、これは旧来のキャピラリーシークエンサーの解析量の100万倍にも相当します。ヒトゲノムのハプロイドあたり総塩基数が約3 Gbpで有ることを考えると、ゲノム総量の100-200倍もの塩基情報が得られるようになりました。
これを用いてゲノム(あるいはエクソン領域)の体細胞変異を網羅的にスクリーニングし、特定のがん種で繰り返し出現するアミノ酸置換変異(非同義変異:nonsynonymous mutations)を探索することも出来ますし、あるいは特定の抗がん剤を使用した際の治療反応性を規定する遺伝子配列異常、さらには浸潤転移・薬剤耐性の直接的な原因となる遺伝子異常を見つけ出すことが出来ます。
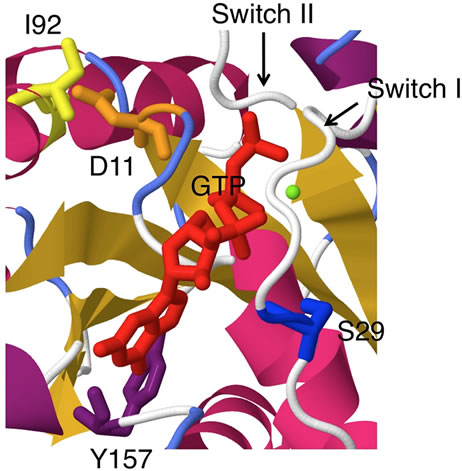
さらに私たちは「次世代シークエンサーによる遺伝子変異探索」と「cDNA発現ライブラリーを用いた機能スクリーニング」の両者を同じ検体に対して行うことで、発がん能があり変異もある遺伝子を効率よく同定することを目指しています。例えばヒトの肉腫細胞株HT1080に対して本解析を行う事で、RAC1という低分子量GタンパクのN92I変異と、別の低分子量GタンパクNRASのQ61K変異を発見しました(PNAS 10:3029)。どちらもマウス線維芽細胞の形質転換を誘導し、明瞭な発がん能が確認されました。興味深いことにHT1080細胞株のNRASをRNAiによってノックダウンしても細胞死は誘導されませんでしたが、RAC1をノックダウンすると明瞭な細胞死が生じ、RAC1(N92I)こそが本がん種の本質的な発がん原因である事が明らかになりました。さらにN92I変異以外にもP29SやC157Yなど様々ながん化変異がRACファミリータンパクには存在する事が明らかになり、がん化RACタンパクが新たな治療標的として重要な事が証明されました。こうして機能アッセイと次世代シークエンサーによる網羅的変異解析とを組合わせて解析することで、私たちは更なる発がん原因の探索に挑んでいます。