トップページ > 研究組織一覧 > 分野・独立ユニットグループ > ゲノム解析基盤開発分野 > 研究プロジェクト
研究プロジェクト
がんゲノムの未解明領域の解明・完全再構成基盤の構築
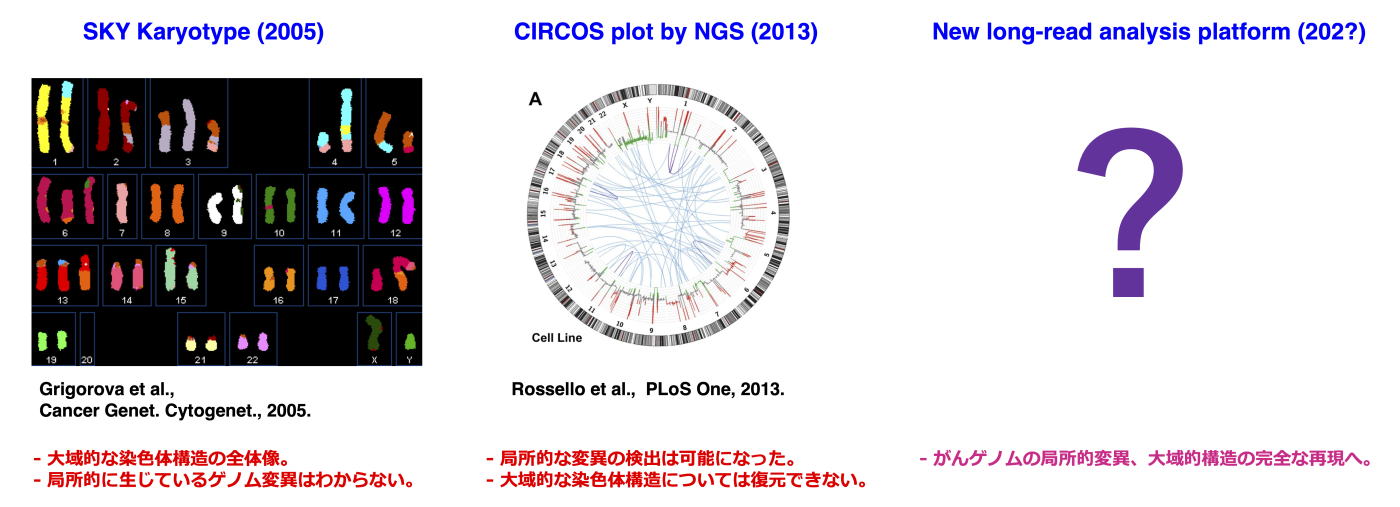
現在、世界中で「全ゲノム解析」に関連した多くのプロジェクトが進められております。全ゲノム解析は、特定の選択をせずに抽出したDNAを高速シーケンサーで読み取り、ゲノム変異を網羅的に探索する技術です。コスト削減のために主要な遺伝子領域のDNAだけを抽出する「ターゲットシーケンス」と異なり、ゲノム全域に生じている変異を網羅的に同定することができるとされております。一方で、ヒトゲノムの多くは「リピート配列」から構成されており、現在主に利用されているショートリードに基づくシークエンスにおいては、多くのゲノム領域の解析が困難・不可能であり、未解明なままとなっているのが実情です。
この状況を打破するために期待されているのが、近年急速に発展しているロングリードシーケンス技術です。この技術により、セントロメアなどの高度リピート領域を含むヒトゲノムの完全配列の決定が可能になりつつあります。しかし、ロングリード技術においても、取得できる配列の長さは10kbpから最大で1Mbp程度に限られ、テロメアの始端から終端まで繋がるゲノムの再構成は依然として困難です。がんゲノムの場合は、複雑な染色体間転座、異数性、サブクローンの問題など、さらに多くの課題が存在します。
私たちの研究室では、中長期的な主要な目標の一つとして、がんゲノムの配列を完全に再構成できる情報解析基盤を開発しています(シン・ゲノム解析基盤と仮称します、図1)。この解析基盤により、まずはがんゲノムを完全な状態で「観る」ということを目指します。さらに、がんゲノム解析のコミュニティにも利用可能な形で提供し、研究と医療において『完全がんゲノム配列』が利用できる世界を創り出すことを思い描いて研究を進めています。
現在、この目標に向けた第一歩として、ロングリードシークエンスを用いた複雑な構造異常解析のための解析アルゴリズムの開発を進めています(Shiraishi et al., Nucleic Acids Research, 2023)。また、ショートリードではほとんど未解明だったセントロメア配列を含むダークマター領域に生じているゲノム変異の解読を世界に先駆けて進めています。
関連ページ:
全ゲノム解析から「シン・ゲノム解析」へ
自律駆動型大規模ゲノムデータ解析基盤の開発
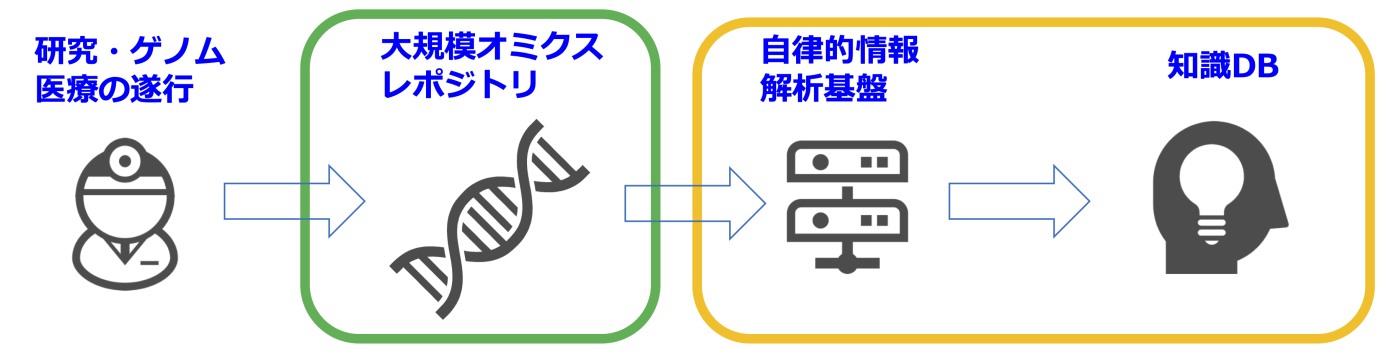
現在、世界中で、研究・医療の目的でゲノムシークエンス解析が盛んに行われています。初めに特定の目的に応じた一次解析から始まりますが、ゲノム元データには直接的な目的を超えた膨大な情報が含まれています。そのため、この元データを世界中の研究者がアクセスできるレポジトリ・データ解析基盤に公開し、再解析を促進することが一般的です。例えば、The Cancer Genome Atlasプロジェクトでは、約10,000件のがん検体のゲノム、トランスクリプトーム、エピゲノムなどオミクスデータが世界中の研究者に公開されており、多角的な再解析を通じてがん研究の大きな進展が達成されました。このようなデータの共有と再活用の流れは、今後も拡大を続けており、世界中でゲノムデータの蓄積・共有が進んでいます。こうした大規模ゲノムデータを効率的に利活用(新たな医学的知見の導出、新規の創薬、データベースの精緻化など)できることはますます重要になるでしょう。
こうした背景の中で、本研究室では、大規模データから可能な限り効率的に「知識」を導出するシステムを開発し、科学・医療にイノベーションを起こすことを目指しています。ここでの「知識」とは、疾患に関連する遺伝的変異や、がんの進展に影響を与える遺伝子異常と薬剤の関係などの情報が集まったデータベースが例として挙げられます(他にもどのような「知識」を体系化できるかを考え続けています)。具体的な取り組みとして、数十万検体規模のトランスクリプトームデータを用いた新たな病的変異のスクリーニング法を開発し(Shiraishi et al., Nature Communications, 2022; Iida et al., bioRxiv, 2024)、これらの医療応用を共同研究者と進めています。また、LLM(Large Language Model)を通じて新しい研究推進の形を模索しています。
関連ページ:
 自律的な知識獲得基盤の概念図
自律的な知識獲得基盤の概念図
がんゲノム研究・医療への貢献
がんゲノム・トランスクリプトームのシークエンスデータを効率的に処理して、様々な解析を行う解析ワークフローの構築を進めております。これまでに、骨髄異形成症候群におけるRNAスプライシング関連の遺伝子変異の発見(Yoshida, Sanada, Shiraishi et al., Nature, 2011)、腎臓がんにおけるゲノム、トランスクリプトーム、メチロームの統合的解析(Sato, Yoshizato, Shiraishi et al, Nature Genetics, 2013)、成人T細胞白血病における新しい分子異常メカニズムの解明(Kataoka, Nagata, Kitanaka, Shiraishi et al., Nature Genetics, 2015)、免疫チェックポイント遺伝子における新規ゲノム異常の発見(Kataoka, Shiraishi et al., Nature, 2016)など数多くのがんゲノム研究に貢献をしてきました。
またその過程で、ベイズ統計理論などを用いた後天的変異の検出(EBCall, Shiraishi et al., Nucleic Acids Research, 2013)、スプライシング変異の網羅的探索手法(SAVNet, Shiraishi et al., Genome Research, 2018; PCAWG Transcriptome Core Group et al., Nature, 2020)、機械学習に基づく変異のパターンマイニング手法(pmsignature, Shiraishi et al., PLoS Genetics, 2015)など、がんゲノム解析のための様々なアルゴリズム・ソフトウェアの開発を行ってきました。
現在、さらに先端的なクラウド・人工知能技術などを取り入れ、効率的なゲノム解析基盤の開発・改良を進めており、国立がんセンター中央病院で進められている全ゲノム患者還元プロジェクトなど数多くのゲノム研究・医療プロジェクトに貢献しています。また、遺伝性腫瘍を専門とする臨床医・研究者と共同で、ロングリードを使った解析ワークフローの開発を進めています(Nakamura et al., npj Genomic Medicine, 2024)。
関連ページ

